CREST Teaching and Research Symposium – April 30, 2011
In spring 2011, the first group of CREST students completed a variety of modeling projects. Several focused on designing drugs to fit in the active site of a protein involved in a cell signaling cascade. Others focused on using a homology model to elucidate the structure of an as yet uncharacterized protein. Some focused on designing a model that would inform ongoing laboratory research. And at least one focused on developing a model and poster that can be used directly in the classroom.
Eleven CREST teams from five institutions in the region gave oral and poster presentations describing their protein modeling projects to an audience of their peers, faculty and researchers at the CREST Research and Teaching Symposium at MSOE on April 30, 2011.
Symposium Program (arranged to create a booklet if printed double-sided)UW-Milwaukee
Bacterial RNA Polymerase: New Insights on a Fundamental Molecular Machine
Students:
- Catherine Dornfeld
- Christopher Hanna
- Jason Slaasted
Faculty Advisor:
- Steven Forst, Ph.D.
Research Mentor:
- Rick Gourse, Ph.D., UW-Madison
PDB file:
- 2o5i.pdb
Presentation:
Abstract:

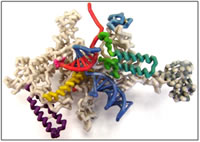
RNA polymerase (RNAP) is an information-processing molecular machine that copies DNA into RNA. It is a multi-subunit complex found in every living organism. Bacterial RNAP contains 6 subunits (ββ’α2ωσ). The ββ’ subunits form several distinct functional channels that accommodate double stranded DNA and the RNA-DNA hybrid as well as the exit channel that guides the growing RNA strand out of the complex and the secondary channel that allows nucleotides to enter the active site. This model focuses on the β’ subunit of Thermus themophilus that contains the highly conserved active site sequence and several structures involved in the catalytic mechanism. The bridge helix (BH) and trigger loop (TL) work together as a “gate” to enhance the catalytic action by facilitating nucleotide addition. In the crystal structure of the RNAP elongation complex (EC) without NTP in the active site the TL (β’ 1236-1265) is unstructured. In the EC crystal structure with a non-hydrolysable nucleotide (AMPcPP) the TL folds into 2 anti-parallel helices (trigger helix, TH) that interact with the adjacent BH to create a 3 helical bundle forming a catalytically active complex. The other structures that are functionally important in the β’ subunit are the “lid” (β’ 525-539) that cleaves the RNA-DNA directing the newly formed RNA out through the exit channel and the “rudder” (β’ 582-602) that helps to stabilize the DNA helix and the RNA-DNA hybrid in the active site channel.£
Mount Mary College
Conserved through the Ages: GATA-1 and Friend of GATA-1(FOG-1) Interactions with DNA
Students:
- Esmeralda Ambriz
- Linnea Esberg
- Nicole Fischer
- Amy Ramirez
Faculty Advisor:
- Colleen Conway, Ph.D.
Research Mentor:
- Michele Battle, Ph.D., Medical College of Wisconsin
PDB files:
- 1y0j.pdb
- 2gat.pdb
Presentation:
Abstract:

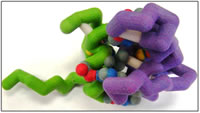
There are six GATA proteins involved in a variety of developmental processes. GATA and FOG are families of zinc finger proteins, which comprise about 3% of the human genome and are often associated with DNA binding activities. The protein interaction of interest is the N-terminus of GATA-1 and the first zinc finger of FOG-1, and these are modeled because they are the only available structures in the protein database. The zinc ion helps to make the tertiary protein structure. GATA proteins are highly conserved and recognize the ‘GATA’ sequence in DNA. Direct physical interaction between the zinc finger transcription factor GATA-1 and cofactor FOG-1 is crucial for erythroid and megakaryocytic development by facilitating transcription. GATA-1 binds to DNA in the promoter and enhancer regions of all erythroid and megakaryote specific genes. Other interaction sites between GATA-1 and FOG-1, which is a 9-zinc fingered protein, include fingers 5, 6, and 9. These interactions ensure normal erythropoiesis and are involved in the later stages of megakaryopoiesis. Without GATA-1/FOG-1 interactions, multiple blood-related diseases result, such as congenital dyserythropoietic anemia or thrombocytopenia. Because of the highly conserved nature of these proteins, determining various relationships and functions of GATA-1 and FOG-1 in embryonic development may lead to clarification of other GATA-FOG relationships. This may also elucidate other complex protein-protein interactions in cells. Using this information might help to focus further questions and design hypotheses to test the function of these in disease and eventually possibly lead to treatments.
Concordia University of Wisconsin
Major Histocompatibility Complex — Class I HLA-A2 and Presentation of HERPES Viral Peptides
Students:
- Joshua Nord
- Lora Rapp
- Nathaniel Wanish
Faculty Advisor:
- Ann McDonald, Ph.D.
Research Mentor:
- Amy Hudson, Ph.D., Medical College of Wisconsin
PDB file:
- 3gso.pdb
Presentation:
Abstract:

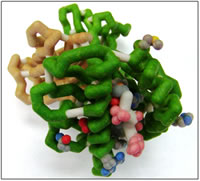
The human immune system has developed the ability to fight many different infectious agents, Cytotoxic T lymphocytes (CTL) are cells of the human immune system that recognize viral-infected cells by binding to portions of viral proteins (peptides) presented on class I major histocompatability complex (MHC) proteins. MHC proteins are located in the cytoplasmic membrane of all nucleated cells and are composed of two protein chains, an alpha (α) chain and a beta-2 microglobulin. The alpha chain is an integral membrane protein consisting of a transmembrane region, as well as three external immunoglobulin domains called α 1, 2, and 3. The base of the peptide-binding cleft is composed of an antiparallel beta-sheet formed between two alpha helices from the alpha-1 and 2 domains. Beta-microglobulin is loosely associated to the α3 domain and important for the stabilization of the class I MHC. Humans without β2-microglobulin will not express class I MHC on the surface of their cells and are prone to recurring viral infections. Herpes viruses establish long-term latent infections achieved by evading class I MHC antigen presentation. Human herpesvirus-7 (HHV-7) produces a unique protein, U21, that blocks expression of human class I MHC, HLA-A, which prevents CTL recognition.
Marquette University
Modeling and Inhibition of VEGF-C/VEGFR2 Active Site
Students:
- Matthew Flister
- Mohamed El Mansy
- Denan Wang
Faculty Advisor:
- Daniel Sem, Ph.D.
Research Mentor:
- George Wilkinson, Ph.D., Children’s Research Institute/Children’s Hospital of Wisconsin
PDB file:
- 2x1w.pdb
Presentation:
Abstract:

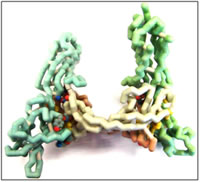
Recent studies have shown that targeting VEGF is a promising anti-cancer treatment since it is known to be responsible for angiogenesis (Argraves and Drake, 2005). Blood supply plays a dual role in tumor growth; it supplies oxygen and nutrients, but also carries chemotherapy and other drugs to the tumor (Folkman, 2002). Tumor vasculature is often leaky and has poor blood flow. VEGF antagonism actually improves blood flow and drug access, but probably also improves oxygenation of the tumor (Folkman et al., 1971). Therefore, inhibiting VEGF binding is likely important in cancer treatment. Studying VEGF inhibition was done mainly with molecular modeling. The active site was characterized with modeling software and VEGF inhibitor targets were designed based on these modeling studies. The designed ligands were optimized using quantum mechanics based computational methods and several homology studies were done to assess the viability of targeting the VEGFR2 protein.
Marquette University
C-Src Kinase Inhibition: A Promising Route in Kidney Cancer Treatment
Students:
- Simon Duri
- Xixi Hong
- Joseph Lustig
- Aleksandra Porebska
Faculty Advisor:
- Daniel Sem, Ph.D.
Research Mentor:
- Ellis Avner, M.D., Medical College of Wisconsin
PDB file:
- 2h8h.pdb
Presentation:
Abstract:

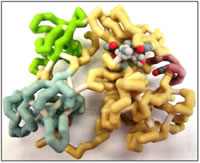
C-Src is involved in a number of signaling pathways that ultimately lead to angiogenesis. Recently derived data leads to the deduction that the most important consequence of increased c-Src activity is promotion of an aggressive phenotype in multiple human tumors. Based on that information, inhibition of c-Src kinase is essential and might be considered a treatment of cancer. There are currently a number of drug molecules targeting c-Src which are in clinical trials. These include Bosutinib which has been shown to significantly reduce the renal growth in cystic bpk mouse models. Drug design can be accomplished with the aid of a number of computer software programs. These help in mapping the active site of the target molecule, which will then be used as a template for possible drug molecules. Minimum energy structures of these molecules can be obtained from programs like Spartan. However, experimental work to establish actual binding constants, solubility and toxicity of chosen drug molecules will have to be carried out.
Marquette University
Drug Design for Inhibition of Extracellular Signal-Regulated Kinase 2 (ERK2)
Students:
- Sam Klingbeil
- Nicole Reiff
- Jay Wagner
Faculty Advisor:
- Daniel Sem, Ph.D.
Research Mentor:
- Ramani Ramchandran, Ph.D., Children’s Research Institute/Medical College of Wisconsin
PDB file:
- 3i60.pdb
Presentation:
Abstract:

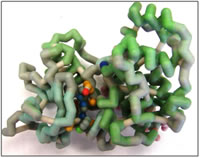
ERK-2, an extracellular signal-regulated kinase, also known as MAPK, mitogen activated protein kinase, is an important protein in the angiogenesis pathway. ERK-2 is vital in controlling the proliferation of vascular endothelial cells. A vascular endothelial growth factor (VEGF) binds its receptor, a tyrosine kinase receptor, starting a signal cascade. In humans, the VEGF receptor recruits phospholipase C gamma (PLC-γ) which is activated by protein kinase C (PKC) via phosphorylation. An active PLC-γ phosphorylates ERK-2, bypassing the normal Ras-Raf-MEK-MAPK pathway (Bellou et al., 2009). The phosphorylated ERK-2 enters the nucleus and phosphorylates, or activates, transcription factors such as Elk-1 and TFIIIB, resulting in cell proliferation (Felton-Edkins et al., 2003).
ERK2 is highly regulated in order to prevent excess endothelial cell proliferation. ERK-2 activity is regulated by the phosphorylation state, typically by using MAPK phosphatases (MKPs) to deactivate ERK-2 and PLC-γ to activate it. DUSP5, a MAPK, contains a phosphatase domain and an ERK2 binding domain. When DUSP5 binds to ERK-2, it dephosphorylates, thus deactivating, ERK-2 using the phosphatase domain. If ERK-2 is not dephosphorylated it remains active leading to excessive cell proliferation. Inhibitors for ERK-2 are a current research topic; Alex M. Aronov et al. at Vertex Pharmaceuticals have developed a drug that inhibits ERK-2, which is currently in Phase 1 clinical trials (Aronov et al., 2009).
Marquette University
The Importance of Understanding DUSP5 for Angiogenesis Prevention

Students:
- Scott Beard
- Dan Jashinsky
- Brandon S. Uhler
Faculty Advisor:
- Daniel Sem, Ph.D.
Research Mentor:
- Ramani Ramchandran, Ph.D., Children’s Research Institute/Medical College of Wisconsin
PDB file:
- 2g6z.pdb
Presentation:
Abstract:

The study of DUSP5 bears a high relevance in the study of disease found in blood vessels (Alonso et al., 2004; Chang and Karin, 2001). The particulars of the study include tumors/growths, angiogenesis that occur within the blood vessels (Pramanik et al., 2009). If we discover how blood vessels are built, we should be able to accordingly know how to destroy them or prevent their growth in the particular cases of disease. Dr. Ramchandran and his team have taken a special interest in researching into battling the development of growths and tumors in the skin of children. Studies are being conducted on zebra fish as a model organism, due to the availability of the fish in addition to the ability to easily view a living vascular system (after 48 hours or so, the fish’s vascular system will be fully developed and fully visible through its clear skin) (Qian et al., 2005; Sumanas et al., 2005). The DUSP5 and Snrk-1 proteins are regulators of cells involved in the vascular mutations of the DUSP5 protein that result in the tumors (Pramanik et al., 2009). A mutation in the DUSP5 protein (e.g. S147P) makes it unable to dephosphorylate the pERK 1/2 protein to ERK 1/2 (Bellou et al., 2009; Aronov et al., 2009). This inability to carry out its function is believed to be the cause of the tumor proliferation in the body. Why does this occur? Phosphorylated ERK1/2 (pERK) activates cell surface tyrosine kinases within the nucleus, which include the epidermal growth factors (Farooq et al., 2001; Aronov et al., 2009). When the ERK cannot be dephosphorylated, it cannot be told to stop activating the growth factors which leads to these tumors. Experiments have shown that the mutated DUSP5 protein is unable to dephosphorylate, since the presence of a mutant DUSP5 shows a constant amount of pERK in the cell with very little ERK (Bellou et al., 2009).
Marquette University
Proposing Ligands and an Active Site in NgBR for Cancer Treatment
Students:
- Geng Lee
- Anna Weber
- Qianhong Zhu
Faculty Advisor:
- Daniel Sem, Ph.D.
Research Mentor:
- Robert Miao, Ph.D., Medical College of Wisconsin
PDB file:
- 2vg1.pdb
Presentation:
Abstract:

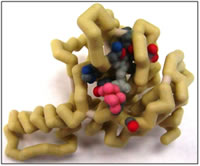
Nogo-B, the primary Nogo isoform, is expressed in blood vessels and binds to the Nogo-B Receptor (NgBR) (Miao et al., 2006). The NgBR protein increases endothelial cell (EC) migration to wherever Nogo-B (soluble protein) is present while decreasing the migrating ability of vascular cells (Oertle et al., 2003; Zhao et a., 2010). Since blood vessels are essential to life, Nogo-B is found in most tissues (Huber et al., 2002; Josephson et al, 2002). The intracellular domain of NgBR can bind to the Farnesyl group in Ras, an oncogene which is an important mediator in the RTK pathway (Miao et al., 2006). Increased interactions between the NgBR and its ligand may cause cancerous cell growth. Experiments in Dr. Robert Miao’s lab at the Medical College of Wisconsin have shown that in zebrafish with their NgBR genetically knocked out, blood vessels were more localized in the central regions of the fish and did not expand outwards as much as in fish with the receptor (Zhao et a., 2010). The same result was seen in mice, in which mice deficient in Nogo-B had fewer vessel formations (Acevedo et al., 2004; Yu et al., 2009. The next experiment that Miao’s lab is interested in is directed to finding the essential domains (active site) of NgBR. They will determine this by developing mutants in the NgBR, then placing the gene back into the zebrafish to observe the changes. Though this is the next step, the active site, structure, and essential domains in NgBR are still unknown. Therefore we used the homologous structure Farnesyl diphosphate synthetase, whose PDB code is 2VG1, as a template. 2VG1 is bounded by a ligand, E,E-farnesyl diphosphate (EE-FPP) and helps make the cell wall for Mycobacterium tuberculosis (Wang et al., 2008). It also helps us to envision how NgBR can bind to a Farnesyl group on Ras. Using a Blast search, the percent sequence homology was found to be 40% (NCBI). We found key amino acids in the active site by defining various distances around the ligand and observing which amino acids were present. Within 6 Å of this chain are seven hydrophobic amino acids — Ile105, Glu107, Gln110, Ser113, Phe114, Ile117, Tyr135, His137 and Gln138. These create a pocket for the hydrophobic tail of Farnesyl through Van de Waal interactions. The other end of Farnesyl contains a nitrogen and an oxygen. Within 6 Å of these groups are Ser249 and Leu251. Ser249 is polar and thus is likely attracted to the polar head of Farnesyl. Leu251 is non-polar, but is shown to be 4.58 Å from the polar head of the ligand. It is possible this non-polar amino acid helps form a pocket for the ligand and stabilizes the attraction.
Marquette University
Design of Soluble Epoxide Hydrolase Inhibitors as Drug Leads
Students:
- Elise Pellmann
- Jay Kim
- Mike Wild
Faculty Advisor:
- Daniel Sem, Ph.D.
Research Mentor:
- John Imig, Ph.D., Medical College of Wisconsin
PDB file:
- 1vg5.pdb
Presentation:
Abstract:

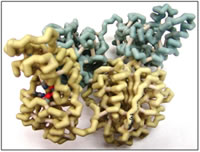
Epoxyeicosatrienoic acids (EETs) are generated from arachidonic acid (ARA) by epoxygenase cytochrome P450. In humans, EETs function as autocrine and paracrine effectors in the cardiovascular system and kidney, where they promote vasodilation and act to inhibit systemic anti-inflammatory response (Spector et al., 2004). The enzyme soluble epoxide hydrolase (sEH) breaks down EETs into dihydroxyeicosatrienoic acids (DHETs). An inhibitor of sEH could therefore act in a physiologically relevant manner by maximizing the amount of EETs in the blood (Imig et al., 2009). A drug-like inhibitor of sEH could have implications in the treatment of cardiovascular disease, kidney disease, and diabetes. sEH is involved in other mechanisms besides EET conversion, and so inhibiting sEH may block other pathways; the physiological significance of this has yet to be evaluated.
In this project, a number of putative sEH inhibitors were designed. Work was based on previous drug design efforts as well as on the three-dimensional structure of the enzyme (Gomez et al., 2004). sEH crystal structures exhibit two domains with distinct activities — the C-terminal domain catalyzes the epoxide hydrolysis reaction for which the enzyme is named, whereas the N-terminal domain exhibits phosphatase activity that is reportedly independent — at least kinetically — of the epoxide reaction. Three amino acids (Asp333, Tyr381 and Tyr465) participate in hydrogen-bonding interactions with inhibitors in the hydrolase active site, which lies in a large, 25 Å-long hydrophobic cavity in the C-terminal domain. Van der Waals interactions with a number of nonpolar residues contribute to hydrolase inhibitor binding. Inhibitor design primarily targeted the hydrolase active site; however, inhibitors of the phosphatase active site were also designed and linked to putative hydrolase inhibitors. These bi-substrate inhibitors are expected to bind sEH with considerable affinity relative to two separate inhibitors due to entropic effects.
UW-Madison
β-Catenin: An Essential Player in Both Cell-Cell Adhesion and Wnt Transcriptional Regulation
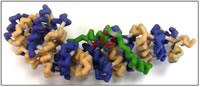
Students:
- Ben Hierlmeier
- Jim Heffernan
- Katie Strobel
- Devan Van Lanen-Wanek
Faculty Advisor:
- Michelle Harris, Ph.D.
Research Mentors:
- Erik Dent, Ph.D., UW-Madison
- Jeff Hardin, Ph.D., UW-Madison
PDB file:
- 1jdh.pdb
Presentation:
Abstract:

β-catenin is a multi-functional protein involved in two essential cellular pathways: cell-cell adhesion and transcriptional regulation. β-catenin contains twelve armadillo repeats capped by a C-helix. An amino acid important for β-catenin’s electrostatic interactions with ligands is Lys435, known as the “charged button”. Given its diverse functions, β-catenin has diverse binding partners. β-catenin’s role in cell-cell adhesion is essential in the early stages of embryogenesis, and defective β-catenin results in inviable embryos. This association is mediated by binding to E-cadherin. β-catenin also mediates events regulated by the Wnt pathway, through its binding of Tcf/Lef family transcription factors via its charged button domain. Wnt signaling is an important regulator of diverse events, including differentiation during embryonic development, and regulated proliferation. Its misregulation by APC, a protein utilized in the Wnt pathway involved in marking β-catenin for degradation, is an important event in colon cancer. Although several binding affinities of β-catenin have been described, several questions remain. In particular, the function of the C-helix is poorly understood. Further studies could help illuminate the role of the C-helix, using various biochemical assays and in vivo analyses in genetic model systems. A physical model would enhance the study of β-catenin’s interaction with its binding partners. An online tutorial would also serve as a valuable teaching tool to illustrate key aspects of the structure of β-catenin that constrain and facilitate its interactions with its binding partners.
UW-Madison
Cdc42 Interacting Protein 4 (CIP4) Involvement in Endocytosis and Membrane Protrusion

Students:
- Genti Gyzeli
- Kelly Mitok
- Corey O’Reilly
Faculty Advisor:
- Michelle Harris, Ph.D.
Research Mentors:
- Jeff Hardin, Ph.D., UW-Madison
- Erik Dent, Ph.D., UW-Madison
PDB files:
- 2efk.pdb
- 2ct4.pdb
- 2ke4.pdb
Presentation:
Abstract:

Endocytosis is a critical process to all living cells. Human Cdc42 interacting protein 4 (CIP4) is known to function in collaboration with other molecules in endocytosis by helping to determine the curvature of the formed vesicle. To do this, certain positively charged residues on the concave surface of the FBAR domain of CIP4 interact with the negatively charged membrane phospholipids. CIP4 is important to the lab we are collaborating with because they have observed it in extending filopodia and lamellipodia of axonal growth cones. This is interesting because the conventionally accepted mechanism that CIP4 interacts with membranes along its concave surface is not consistent with our research that shows this protein is important for protrusion. CIP4-induced filopodial and lamellipodial protrusions would, however, be consistent with it interacting with the membrane along its convex surface also. This potentially novel function of CIP4 is important because it could add to our understanding of axon growth and neuron migration in prenatal nervous system development in humans. In our model of human CIP4 we are focusing on both the positively charged residues on the concave surface of the FBAR domain that have been shown to be important in endocytosis and the positively charged residues on the convex surface that may be important in protrusion. Further research could include carrying out point mutation studies of the positively charged residues on the convex surface of the FBAR domain to assess residues important in filopodia and lamellipodia protrusion.