Molecular Pharmacology Poster Session – April 10, 2012
The Molecular Pharmacology students at Concordia University Wisconsin School of Pharmacy working with Dr. Dan Sem explored the molecular mechanism of drug action. Teams of students met with preceptors (pharmacist mentors) to discuss interesting clinical cases in which a prescribed drug resulted in either unwanted side effects or drug-drug interactions. Each team then explored one of these drug stories, developing a case study, historical background on drug development and the mechanism of drug action. The project involved writing a group paper, creating a 3D model of the protein using Jmol and 3D printing technology, and presenting a poster at the poster session.
Protein models depict the drug docked with the target protein; additional models were subsequently generated, showing a close up of the active site of the protein with the drug docked using magnets. These models are available for loan through the MSOE Model Lending Library. Contact (crestprogram@gmail.com) for information about borrowing this collection.
Glucocorticoid Receptor with Bound Fluticasone Furoate
Poster Development Students:
- Allison Dembeck
- Joseph Dvorak
- Stacy Menzer

Model Development Students:
- James Lokken
- Bores Rempel
- Tristin Sabin
Structure:
- Protein: Glucocorticoid receptor
- Drug: Flutacasone furoate (active component of Veramyst Nasal Spray)
- PDB file: 3cld.pdb
Presentation:
Abstract:
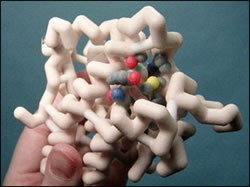 The Glucocorticoid Receptor (GR) is expressed in most mammalian cells and regulates genes controlling development, metabolism, and immune response. Because the receptor gene is expressed in several forms, it has pleiotropic effects in different parts of the body. GR is located in the cytoplasm and is activated when it binds to a type of steroid called a glucocorticoid, with its primary mechanism of action being the regulation of gene transcription. The activated GR complex up-regulates the expression of anti-inflammatory genes and helps regulate the cytochrome P450 (CYP450) family of metabolic enzymes, which are responsible for the metabolism of most drugs. Interactions involving CYP450 enzymes can result in non-therapeutic levels of drugs or even toxicity, as depicted by the potential interaction between fluticasone furoate (FF) and ritonavir. FF is a glucocorticoid with a stronger binding affinity to GR than many other glucocorticoids, because of a few unique binding properties. FF contains a 17 alpha furoate ester which occupies the lipophilic 17 alpha pocket on GR more fully, which may account for the enhanced GR binding of fluticasone furoate as compared with dexamethasone or earlier fluticasone molecules. Within the 17 alpha pocket there are a number of strong van der Waals interactions which accounts for the rapid association and slow dissociation from GR. Also, the 3-keto, 11β hydroxyl, and the 17β fluoromethylthio fluorine groups of FF participate in hydrogen bonding with several key amino acid side chains of GR. These properties are what give FF its affinity for GR.
The Glucocorticoid Receptor (GR) is expressed in most mammalian cells and regulates genes controlling development, metabolism, and immune response. Because the receptor gene is expressed in several forms, it has pleiotropic effects in different parts of the body. GR is located in the cytoplasm and is activated when it binds to a type of steroid called a glucocorticoid, with its primary mechanism of action being the regulation of gene transcription. The activated GR complex up-regulates the expression of anti-inflammatory genes and helps regulate the cytochrome P450 (CYP450) family of metabolic enzymes, which are responsible for the metabolism of most drugs. Interactions involving CYP450 enzymes can result in non-therapeutic levels of drugs or even toxicity, as depicted by the potential interaction between fluticasone furoate (FF) and ritonavir. FF is a glucocorticoid with a stronger binding affinity to GR than many other glucocorticoids, because of a few unique binding properties. FF contains a 17 alpha furoate ester which occupies the lipophilic 17 alpha pocket on GR more fully, which may account for the enhanced GR binding of fluticasone furoate as compared with dexamethasone or earlier fluticasone molecules. Within the 17 alpha pocket there are a number of strong van der Waals interactions which accounts for the rapid association and slow dissociation from GR. Also, the 3-keto, 11β hydroxyl, and the 17β fluoromethylthio fluorine groups of FF participate in hydrogen bonding with several key amino acid side chains of GR. These properties are what give FF its affinity for GR.
Renin with Bound Aliskiren (Tekturna)
Poster Development Students:

- Carissa Fuchs
- Kayci Gruening
- Stacy Lamp
- Erika Niznik
- Cassandra Roberts
- Andrew Russell
Model Development Students:
- Adam Huth
Structure:
- Protein: Renin
- Drug: Aliskiren (Tekturna)
- PDB file: 2v0z.pdb
Presentation:
Abstract:
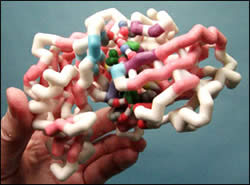
Aliskiren is a novel drug that inhibits the Renin-Angiotensin-Aldosterone System (RAAS) by inhibiting the renin protein. This is the first FDA approved drug that inhibits RAAS at the renin level; other drugs inhibit RAAS at the angiotensin level. Aliskiren binds to the active site of renin and inhibits its enzymatic activity of converting angiotensinogen into angiotensin I. Aliskiren is important because it can lower blood pressure and protect the kidneys of diabetic patients through this unique mechanism of action, which allows it to be used in patients who have failed standard therapies. Because it was rationally designed by medicinal chemists, Aliskiren is a safe drug that specifically binds to renin. In order to effectively design a novel inhibitor for renin the binding pocket was divided into 6 pockets. The chemists discovered that the S3 pocket had a sub pocket which was unused by the substrate. Once they utilized this sub pocket they found it to provide a critical role in specificity. There are over 30 amino acids involved in binding Aliskiren to renin’s binding pocket. The amino acids that are involved in van der Waals interactions are just as critical as the 11 amino acids involved in hydrogen bonds. Due to the number of amino acids involved in binding, only the atoms involved in hydrogen bonding are displayed in the model along with a phenylalanine that is involved with π stacking with Aliskiren. The crystal structure of renin allowed the chemists to design the transition state analog Aliskiren.
Leucine Transporter (LeuT) in Complex with Sertraline (Zoloft)
Poster Development Students:

- Danielle Kroll
- Katie Morawski
- Maria Parish
- Ryan Perry
Model Development Students:
- Christopher Miller
- Matthew Pivec
Structure:
- Protein: Leucine transporter
- Drug: Setraline (Zoloft, Lustral)
- PDB file: 3gwu.pdb
Presentation:
Abstract:
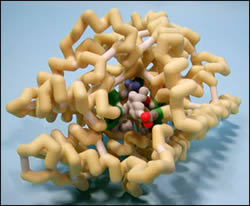
3GWU shows the selective serotonin reuptake inhibitor sertraline bound to the active site of the leucine transporter. This transporter is responsible for recycling synaptic serotonin back into the presynaptic neuron from the synaptic cleft. When the neurotransmitter remains in the cleft, more can bind to postsynaptic neurons to transmit appropriate signals. If this transporter is supersaturated for an extended period of time, an overload of serotonin can create a significant profile of potentially deadly symptoms. Since serotonin is mostly transmitted via neurons in the brain, both the efficacy and toxicity of sertraline and medications like it are mediated via the central nervous system. These toxic symptoms include elevated heart rate and blood pressure, as well as hyperthermia, among others. This drug would be effective in people who metabolize serotonin in the cleft at a faster rate, or for people who do not release as much serotonin into the cleft. The molecular interaction between sertraline and the leucine transporter is important because the pocket is completely occupied by sertraline, forcing the neurotransmitter elsewhere (ie across the synapase to the postsynaptic neuron). The pocket is made up of Phe253, Asp404, Leu400, Asp401 , Ala319, Phe320, Arg30.
Angiotensin-1 Converting Enzyme Bound to Lisinopril
Poster Development Students:

- Gail Kingsbury
- Lexiee Carpenter
- Patrick Tierney
Model Development Students:
- Jennifer Dippel
- Sarah Hunt
- Whitney Hendrickson
Structure:
- Protein: Angiotensin-I converting enzyme (ACE)
- Drug: Lisinopril (Prinivil, Zestril)
- PDB file: 1o86.pdb
Presentation:
Abstract:
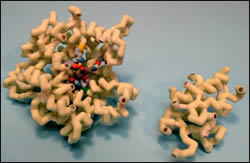
Angiotensin-converting enzyme (ACE) plays a critical role in the regulation of blood pressure within the body. Aldosterone, a steroid hormone secreted from the adrenal cortex, is also an essential component in blood pressure regulation. The production and secretion of aldosterone relies on the interaction of angiotensin II with angiotensin II type 1 (AT1) receptors of the adrenal cortex, and on vascular smooth muscle. This interaction on vascular smooth muscle increases blood pressure through vasoconstriction. ACE is responsible for catalyzing the production of angiotensin II from its inactive precursor angiotensin I, allowing the synthesis and secretion of aldosterone to cause vasoconstriction, and increased blood pressure.
Inhibitors of ACE are an effective drug therapy for hypertension, heart disease and diabetic neuropathy. Lisinopril is one of the 11 ACE inhibitors that is currently FDA approved within the U.S. The binding interactions between lisinopril and ACE play a crucial role in preventing the conversion of angiotensin I into angiotensin II, causing inhibition of the vascular smooth muscle resulting in reduction of high blood pressure. ACE is a Zn2+-dependent dipeptidyl carboxypeptidase composed of two sub-domains that form an ellipsoid structure, with a deep central cavity containing the active site for lisinopril to bind. Both the N- and C-terminal domains have protease activity, but the C-terminal domain plays a larger role in blood pressure regulation. The zinc ion is an important catalytic component in ACE. This ion is bound deep within the active site of ACE, and aids in the hydrolysis of peptide bonds containing hydrophobic amino acids. This zinc-binding motif binds via metal covalent bonds to His383, His387, and Glu411 of ACE. The carboxyalkyl carboxylate group located between the phenylpropyl group and the lysine of lisinopril binds via a hydrogen bond to Glu384 of ACE, near the Zn2+-binding motif. The lysine chain of lisinopril binds to Glu162 of ACE via hydrogen bonding into the S1’ subsite. The C-terminal proline carboxyl group of lisinopril binds to Lys511 and Tyr520 via hydrogen bonding into the S2’ subsite of ACE.
HMG-CoA Reductase with Bound Atorvastatin (Lipitor)
Poster Development Students:

- Kristin Blunt
- Sam Fischer
- Michelle Lynn
- Isaac Olstad
- Huan Phan
Model Development Students:
- Linnea O’Toole
- Elizabeth Weinfurtner
Structure:
- Protein: HMG-CoA reductase
- Drug: Atorvastatin (Lipitor)
- PDB file: 1hwk.pdb
Presentation:
Abstract:
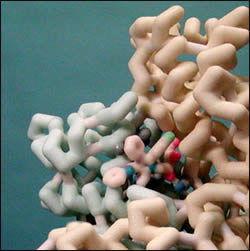
With the high prevalence of hyperlipidemia in the United States, we wanted to explore further one of the most popular classes of cholesterol medications, HMG-CoA reductase inhibitors, or statins. In particular we looked at atorvastatin, which recently became available generically. Our team researched HMG-CoA reductase, a key enzyme involved in the production of cholesterol. Atorvastatin, along with the other statin medications, inhibits this enzyme via competitive inhibition. In other words, the drug looks similar enough to the natural substrate, HMG-CoA, that it binds to reductase and thus blocks it from binding to HMG-CoA, halting cholesterol synthesis. The enzyme is comprised of four identical chains, with four binding regions. Our model shows two of these chains and how they form one of the binding pockets. We also display the drug itself bound in the pocket, and the pertinent interactions that hold it in place.
By inhibiting HMG-CoA reductase, statins not only limit cholesterol synthesis, but also other products such as coenzyme Q10 and heme-A. It is believed that the decrease in these other products may be the cause of statin-related muscle weakness, or myopathy. Further research would be needed to find a medication that could inhibit cholesterol synthesis without blocking other products.
Spironolactone Bound to the Mineralocorticoid Receptor
Poster Development Students:

- Emily Aycock
- David Baszynski
- Sam Fraser
- Spencer Gull
- Morgan Hamm
Model Development Students:
- Jackie Claypool
- Hoda Saedi
Structure:
- Protein:Mineralcorticoid receptor
- Drug: Spironolactone (Aldactone)
- PDB file: 2ab2.pdb
Presentation:
Abstract:
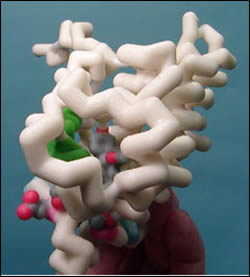
The mineralocorticoid receptor (MR) plays a major role in controlling blood pressure. This receptor has a high affinity for mineralcorticoids (aldosterone is an example), which once bound, result in the activation of Na+/K+ pumps. This process leads to an increase in plasma volume, resulting in a rise in blood pressure. Spironolactone is a synthetically designed, potent antagonist of the mineralocorticoid receptor. Its antagonistic character is due to a C17 γ-lactone. Various substituents at different positions of the steroid skeleton were modified during development to increase potency. Over the last 30 years, spironolactone has remained one of the most widely used mineralocorticoid receptor antagonists. Its use ranges from ascites, congestive heart failure, edema, and hypertension to hypokalemia, primary aldosteronism, and acne vulgaris.
This model depicts the antagonistic spironolactone bound to the MR. MR has three major functional domains: a central DNA binding domain, an N-terminal activation domain, and a ligand binding domain which contains the C-terminal activation function-2 helix (AF-2). To be activated, MR undergoes a conformational change in which AF-2 must be aligned to recruit transciption coactivators. This requires multiple interactions. The loop preceding the AF-2 is stabilized via hydrogen bonds between Asn770 and Ser767 on helix 3 and Glu955 on the loop. Additionally, helix 3 is stabilized via hydrogen bonding with Thr945. Weakened bonding potentials between the lactone keto moiety of spironolactone and Asn770, do not allow for proper positioning of the AF-2, enabling spironolactone to bind to MR without activating it.
Human Serum Albumin Complexed with Warfarin
Poster Development Students:

- Tamara Brandl
- Angela Colella
- Carissa Drewa
Model Development Students:
- Jeremy Bartelt
- Travis Krans
- Benjamin Mattila
Structure:
- Protein: Human serum albumin
- Drug: Warfarin (Coumadin, Jantoven, Marfarin)
- PDB file: 2bxd.pdb
Presentation:
Abstract:
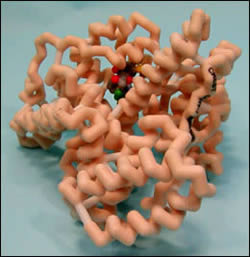
Warfarin is a common anticoagulant medication that is part of many therapy regimens for people throughout the world. Warfarin’s effect is life saving for multitudes of people suffering from numerous disease states. However, since its principle effect is in elongating the body’s ability to clot it can also be quite lethal if not monitored properly. Therefore, fully understanding this medication and its interactions is imperative for all healthcare professionals. This study seeks to elucidate the pharmacological properties behind the interaction between warfarin and phenytoin; which is a drug used frequently to treat seizures.
In the human body warfarin binds to Human Serum Albumin (HSA) proteins. This binding occurs at the IIA domain and consists of both hydrophobic and electrostatic interactions. These interactions are expected to help modulate the drugs activity in the body. Unfortunately, phenytoin (as well as some other drugs) can bind to the same site on HSA as warfarin. This competitive binding of multiple drugs, specifically phenytoin in this case, for the same binding site can result in unpredictable fluctuations in warfarin levels which can be lethal. By understanding this interaction and knowing what may occur, healthcare professionals can make the necessary adjustments in warfarin dosing to ensure that a patients’ warfarin level will remain within the therapeutic range.
Donepezil (Aricept) Complexed with Acetylcholinesterase
Poster Development Students:

- Lara Johnson
- Tiffany Palm
- Melodie Romanowich
Model Development Students:
- John Dalton
- JiYoung Kim
- Steve Scholzen
Structure:
- Protein: Acetylcholinesterase
- Drug: Donepezil (Aricept)
- PDB file: 1eve.pdb
Presentation:
Abstract:
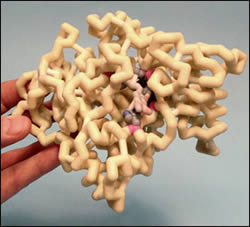
Acetylcholine (ACh) is one of the major neurotransmitters involved in the pathophysiology of Alzheimer’s disease (AD). A decrease in levels of ACh in the brain is thought to be involved in some of the cognitive symptoms of AD, specifically memory loss. There are currently only a few drugs on the market which are approved to treat Alzheimer’s disease, one of them being donepezil. Donepezil is an N-benzylpiperidine-based acetylcholinesterase (AChE) inhibitor that has three distinctive portions of the molecule. The structure is made up of a dimethoxyindanone portion, a piperidine portion, and a benzyl ring. All three segments of the molecule have specific interactions within the binding site of AChE. Donepezil is a reversible AChE inhibitor, which binds to the active site of the enzyme that degrades ACh and thus prevents the hydrolysis of it. This results in an increased concentration of ACh in the synapses available for neurotransmission. While this drug can improve some of the cognitive symptoms of AD, it does not treat the underlying cause of the disease.
Hydrochlorotiazide (Aquazide H) Complexed with AMPA Subunit GluR2
Poster Development Students:

- Rebecca Rypel
- Michael Tilkens
- Kristin Wejrowski
Model Development Students:
- Erin Gehrke
- Kristel Mtsig
Structure:
- Protein: AMPA subunit GluR2
- Drug: Hydrochlorothiazide (Aquazide H, Carozide)
- PDB file: 3ijx.pdb
Presentation:
Abstract:
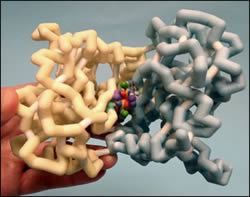
Hydrochlorothiazide (HCTZ) has exclusively been associated with the sodium-chloride co-transport system and its effects as a diuretic and hypertension agent. However recent research is shifting this focus of HCTZ to its effects on a new target, AMPA receptors. AMPA receptors have distinct ionotropic glutamate-excitatory functions in post-synaptic areas of the brain that lead to important neurophysiologic processes. AMPA receptors have important implications due to their association with diseases including Huntington’s, Parkinson’s, and Alzheimer’s. Many patients with these health issues may be treated with modulators of AMPA receptors, such as HCTZ. Our model represents the binding of HCTZ as a dimer to the iGluR2 subunit of AMPA receptors. HCTZ is held into the binding pocket of the iGluR2 subunit via two hydrogen bonding interactions; one between the 4-nitrogen on HCTZ and Lys218 residue of the receptor and the other between the Ser108 residue of the receptor and the sulfonamide group of HCTZ. Another important feature is the hydrogen substitute on the 3-carbon rotating HCTZ which allosterically modulates the receptor, leaving the B subunit open for water molecules to fill the space, rendering the site hydrophilic. This conformational change in the receptor could be the key in drug development as it relates to glutamate receptor binding affinity. This action results in an increase in neuronal activity of the hippocampus, which is accomplished by inhibiting the desensitization of the receptor, which allows a prolonged excitatory state, increased neuron firing, and is therefore a potential drug target for treating neurological diseases involving AMPA receptors.
Celecoxib (Celebrex) Complexed with COX-2
Poster Development Students:

- Dustin Ableidinger
- Jason Barnes
- Philip Frye
Model Development Students:
- Elizabeth Laubach
- Jennifer Ditter
Structure:
- Protein: COX-2
- Drug: Celecoxib (Celebrex)
- PDB file: 3ln1.pdb
Presentation:
Abstract:
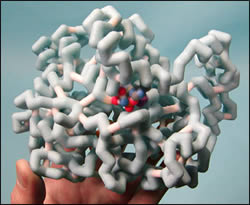
Pain management is often an important part of patient care as it is very real and relevant to a patient’s mental state throughout their disease or recovery process. Typical over the counter (OTC) medications are not always sufficient to appropriately manage pain control, nor does their OTC status render them safe for long term use. Many OTC medications are targeted to bind to proteins to block the inflammation process such as COX-2, but in doing so, evoke side effects because they are able to bind to other proteins as well, such as COX-1. One option patients may receive is an anti-inflammatory medication that binds specifically to COX-2. Celecoxib (Celebrex) is such a drug and offers potent anti-inflammatory effects without unwanted symptoms in the stomach and intestines because it does not bind to COX-1. Our model attempts to show how celecoxib binds to COX-2 and what makes the relationship between the drug and protein unique. For example, we have highlighted a particular amino acid, valine, in violet as this represents a key difference in structure between COX-2 and COX-1. We have also shown the drug itself and the way it fits into the COX-2 protein by coloring the majority of COX-2 in light blue. While celecoxib offers pain management with fewer side effects, other drugs that are similar to it have caused serious problems, and new research is being conducted to try and find medications similar to celecoxib.
2-Bromoacetoxy-benzoic Acid (Aspirin Analogue) Complexed with Prostaglandin H2 Synthase (Cyclooxygenase)
Poster Development Students:

- Amanda Costopoulos
- MMax Grider
- Tyler Nickel
Model Development Students:
- Kate Fiacchino
- Janelle Juul
Structure:
- Protein: Prostaglanding H2 synthase (cyclooxygenase)
- Drug: 2 bromoacetoxy-benzoic acid (aspirin analogue)
- PDB file: 1pth.pdb
Presentation:
Abstract:
The cyclooxygenase enzyme , which is also known as prostaglandin H2 synthase (PGHS) or prostaglandin endoperoxide synthetase (PES), has a role in the formation of mediators such as prostaglandins, prostacyclins and thromboxanes. These mediators are collectively known as eicosanoids. The first and committed step in the production of prostaglandins occurs in the cyclooxygenase active site and involves the addition of two oxygen molecules to arachindonate to form hydroperoxy endoperoxide prostaglandin G2 (PGG2). PGG2 is then reduced to prostaglandin H2 (PGH2) in the peroxidase active site. The result of this pathway is the formation of prostaglandins, prostacyclins, and thromboxanes PGH2. These chemical mediators are involved in numerous physiologic processes like platelet activation, inflammation, pain and fever. Aspirin is used to reduce the effect of these chemical mediators and their physiologic processes within the human body.
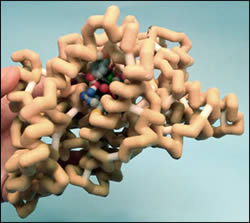
This model of PGHS depicts the well-known mechanism of action of aspirin. The model exemplifies cyclooxygenase inactivated by the aspirin analogue 2-bromoacetoxy-benzoic acid. Acetylation by this analogue stops activity of cyclooxygenase by steric blockage of the active site. In this bromo-inactivated structure, Ser-530 is bromoacetylated (colored in dark slate gray, bromine atom in purple) and a molecule of salicylate (dark slate grey) is bound. The salicylate molecule interacts with the neutral, nonpolar amino acids Val349, Ala527, Leu352 (colored in CPK). These neutral, nonpolar amino acids are distinguished by a hot pink backbone. The salicylate molecule also interacts with the neutral Tyr385 and basic Arg120 amino acids (colored in CPK). These polar amino acids are distinguished by an orange backbone. The peroxidase sites of cyclooxygenase contain a heme (colored in lime green). The iron in the center of the heme is coordinated by His 207 and His 388 (colored in green). Tyr385 is an important catalytically active amino acid residue (colored in teal). Aspirin employs its effects through this selective acetylation of serine 530.
Imatinib (Gleevec) Complexed with Tyrosine Protein Kinase ABL2
Poster Development Students:

- Jacklyn Ekern
- Jason Wettengel
- James Cruikshank
Model Development Students:
- Patrick Pentek
- Jerome Mauro
Structure:
- Protein: Tyrosine-protein kinase ABL-2
- Drug: Imatinib (Gleevec)
- PDB file: 3gvu.pdb
Presentation:
Abstract:
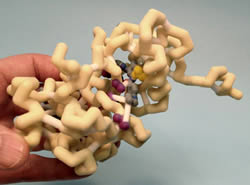
Imatinib mesylate (Gleevec) was the first therapy to specifically target the ABL protein kinase portion of the BCR-ABL oncogene protein which is associated with leukemia. Two imatinib molecules are bound to BCR-ABL, one in the active ATP binding site (ABL2) and the other at a remote site (ABL1). Imatinib inactivates the protein, inhibiting cell proliferation. The kinase domain of the complex consists of two lobes. The smaller N-terminal lobe consists of a five stranded beta sheet and a prominent helix. The bigger C-terminal lobe is composed primarily of helices. The two lobes are connected by a hinge that allows rotation of the two lobes upon ATP and substrate binding. The ATP binding site is located in a pocket created from portions of each lobe. Imatinib binds in an extended conformation, with the pyridylpyrimidine moiety trans to methylbenzene and benzamide rings. Hydrogen bonding occurs between imatinib and Ile406, His407, Asp427, Glu332, Thr361, and Met364. In the inactive conformation, Asp427 points away from the ATP binding site and Phe428 points towards the ATP binding site producing an aromatic stacking interaction with imatinib. Rotation to the active conformation has Asp427 pointing inward and Phe428 pointing outward, interacting with hydrophobic parts of imatinib. Lys317, Met336, Leu416, Leu294, Val302, and Tyr299 form additional hydrophobic interactions. Thr361 and Glu332 are gatekeeper residues that hold imatinib in the correct position via hydrophobic interactions and hydrogen bonding to keep the complex active. Glu332 utilizes resonance to create a stable bond with imatinib in the active conformation. Phe428 and the pyrimidine ring are parallel, creating Pi interactions for a stable bond between imatinib and ABL2 complex..