The 2018-2019 CREST Conversation focused on the science of Christine Dunham, winner of the 2019 ASBMB Young Investigator Award. The Dunham laboratory studies the bacterial ribosome, with special emphasis on the regulation of protein synthesis in response to stress.
Campbell University (Buies Creek NC)
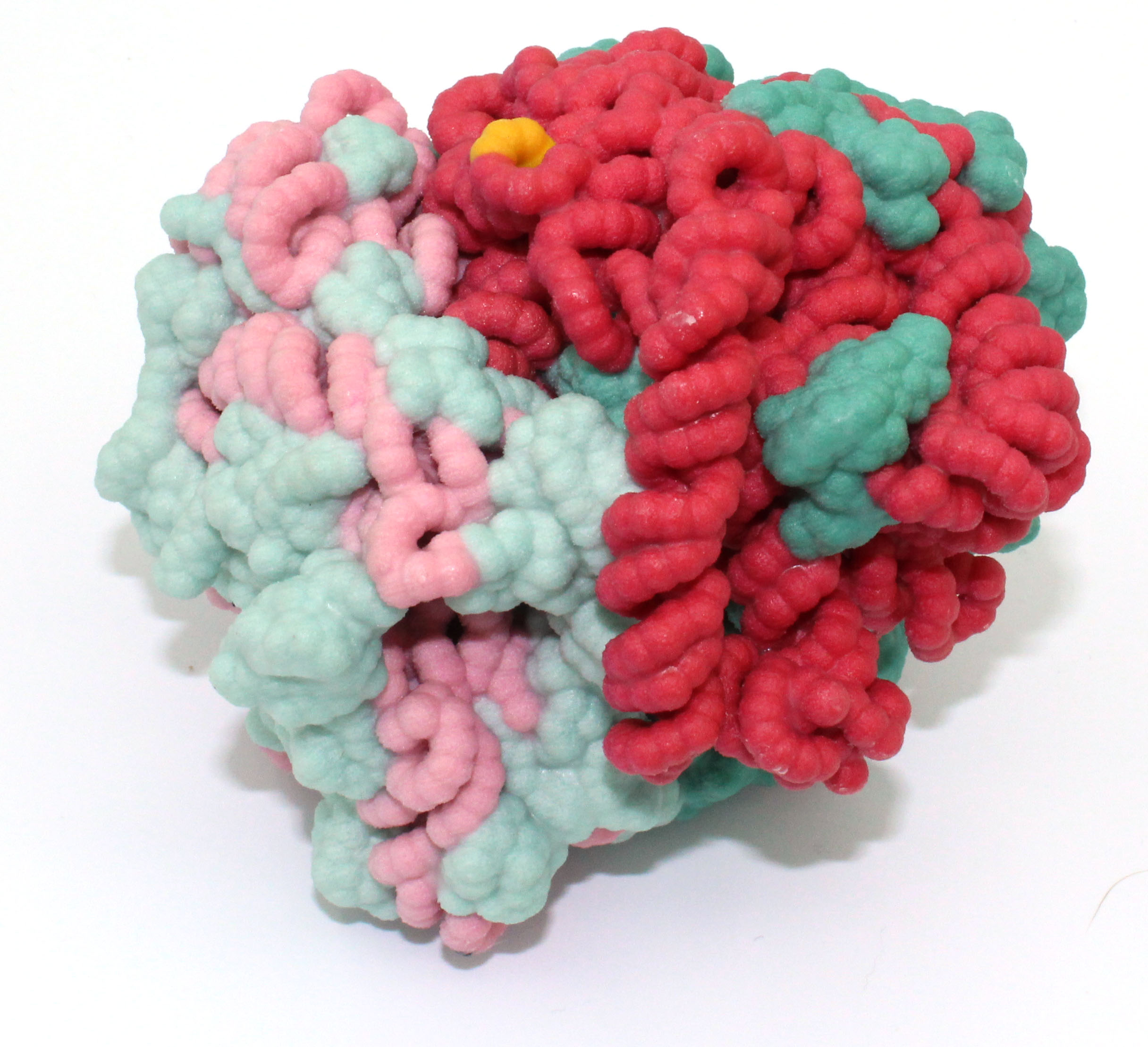
A Physical Model of Mycobacterium tuberculosis toxin MazF-mt6 Illustrates the Catalytic Residues Needed for 23S rRNA Cleavage via a Proton-Relay Mechanism
Aaron Dobbins, Joey Phillips, Cayla Herm and KayLee Russell
Faculty Advisor: Karen Guzman, Ph.D.
PDB file: 5UCT.pdb
Primary Citation: Hoffer, ED, Miles, SJ, Dunham, CM (2017) The structure and function of Mycobacterium tuberculosis MazF-mt6 toxin provide insights into conserved features of MazF endonucleases. J Biol Chem, 292(19): 7718-7726.
Abstract: Toxin-antitoxin systems, which are commonly found in prokaryotes, regulate cell growth in response to environmental stressors. When the labile antitoxin is degraded, the toxin is released to exert its inhibitory effects. The Mycobacterium tuberculosis toxin, MazF-mt6, is a small globular protein composed of two β-sheets and three α-helices. This protein regulates growth by cleaving the 23S rRNA at helix 70 on the large 50S ribosomal subunit, thus inhibiting translation. The 3D structure of MazF-mt6 was recently solved  by the lab of Dr. Christine Dunham. Their work demonstrated that Asp-10, Arg-13, and Thr-36 are critical residues needed for cleavage within the sequence 5’ UU↓CCU 3’ located at positions 1939 to 1943. These catalytic residues perform endoribonuclease cleavage through a proton-relay mechanism, where threonine stabilizes the transition state. Lending support to the location of the active site, a negatively charged sulfate ion, which can mimic the negatively charged phosphate backbone of RNA, is found bound adjacent to the residues that appear to participate in the cleavage reaction. The crystal structure also demonstrates interactions between the α4 helices of separate monomers, supporting previous investigations that indicate the toxin functions as a homodimer. We have designed a physical model to illustrate the 3 catalytic residues of the active site, as well as the bound sulfate ion. Hydrophobic interactions between the residues in the α4 helices of the monomers are also illustrated.
by the lab of Dr. Christine Dunham. Their work demonstrated that Asp-10, Arg-13, and Thr-36 are critical residues needed for cleavage within the sequence 5’ UU↓CCU 3’ located at positions 1939 to 1943. These catalytic residues perform endoribonuclease cleavage through a proton-relay mechanism, where threonine stabilizes the transition state. Lending support to the location of the active site, a negatively charged sulfate ion, which can mimic the negatively charged phosphate backbone of RNA, is found bound adjacent to the residues that appear to participate in the cleavage reaction. The crystal structure also demonstrates interactions between the α4 helices of separate monomers, supporting previous investigations that indicate the toxin functions as a homodimer. We have designed a physical model to illustrate the 3 catalytic residues of the active site, as well as the bound sulfate ion. Hydrophobic interactions between the residues in the α4 helices of the monomers are also illustrated.
Grand View University
Killer Protein HigB and the Inhibition of Translation
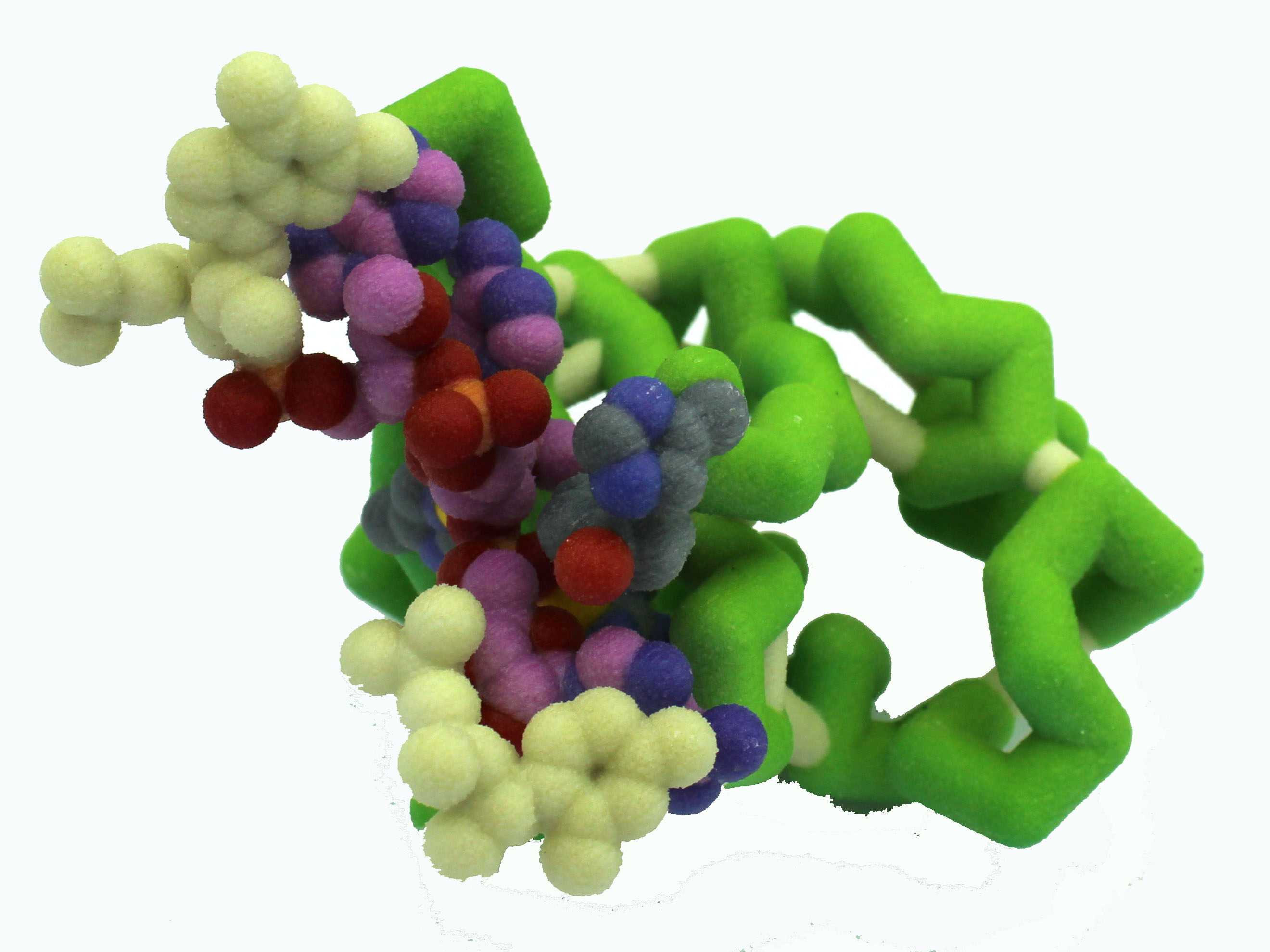
Kami Wheelock, Allison Rupert, Tom Le, Antonio Santa-Maria, Elma Omanovic and Cassandra Corley
Faculty Advisors: Bonnie Hall, Ph.D. and Laura Salazar, Ph.D.
PDB file: 4ZSN.pdb
Primary Citation: Killer Protein HigB and the Inhibition of Translation
Abstract: Toxin-antitoxin systems play a large role in allowing bacteria to survive harsh environmental conditions. The killer protein HigB is one such toxin. During environmental stress, HigB inhibits translation in bacteria by attaching to the ribosome and cleaving mRNA. Dr. Christine Dunham’s laboratory has generated the 4ZSN crystal structure of  the 70s wild type ribosome complexed with the HigB toxin. The 70s ribosome is also bound to an mRNA containing an AAA codon. HigB binds to the aminoacyl site in the ribosome, and cleaves between two of the A nucleotides. The cleavage requires four amino acids: His54, Tyr91, Arg73, and His92. We have created a model highlighting how these amino acids interact with the mRNA that will be cleaved. The model shows the hydrogen bond formed between Arg73 and the mRNA backbone, stabilizing the phosphate group and aligning HigB properly with the mRNA. His54 initiates cleavage of the mRNA by attacking an -OH group on the sugar phosphate backbone. Elucidating how mRNA is cleaved by the killer protein HigB in times of stress may lead to a better understanding of why some bacterial infections are so difficult to eradicate.
the 70s wild type ribosome complexed with the HigB toxin. The 70s ribosome is also bound to an mRNA containing an AAA codon. HigB binds to the aminoacyl site in the ribosome, and cleaves between two of the A nucleotides. The cleavage requires four amino acids: His54, Tyr91, Arg73, and His92. We have created a model highlighting how these amino acids interact with the mRNA that will be cleaved. The model shows the hydrogen bond formed between Arg73 and the mRNA backbone, stabilizing the phosphate group and aligning HigB properly with the mRNA. His54 initiates cleavage of the mRNA by attacking an -OH group on the sugar phosphate backbone. Elucidating how mRNA is cleaved by the killer protein HigB in times of stress may lead to a better understanding of why some bacterial infections are so difficult to eradicate.
Hamline University
Silvia Toledo, Ashley Robinson, Jaxon Renn, Caleb Rosenthal, Maddy Bygd and Dami Ademola-Green
Faculty Advisor: Betsy Martinez-Vaz, Ph.D.
Aminoglycoside-resistance rRNA Methyltransferase A (NpmA)
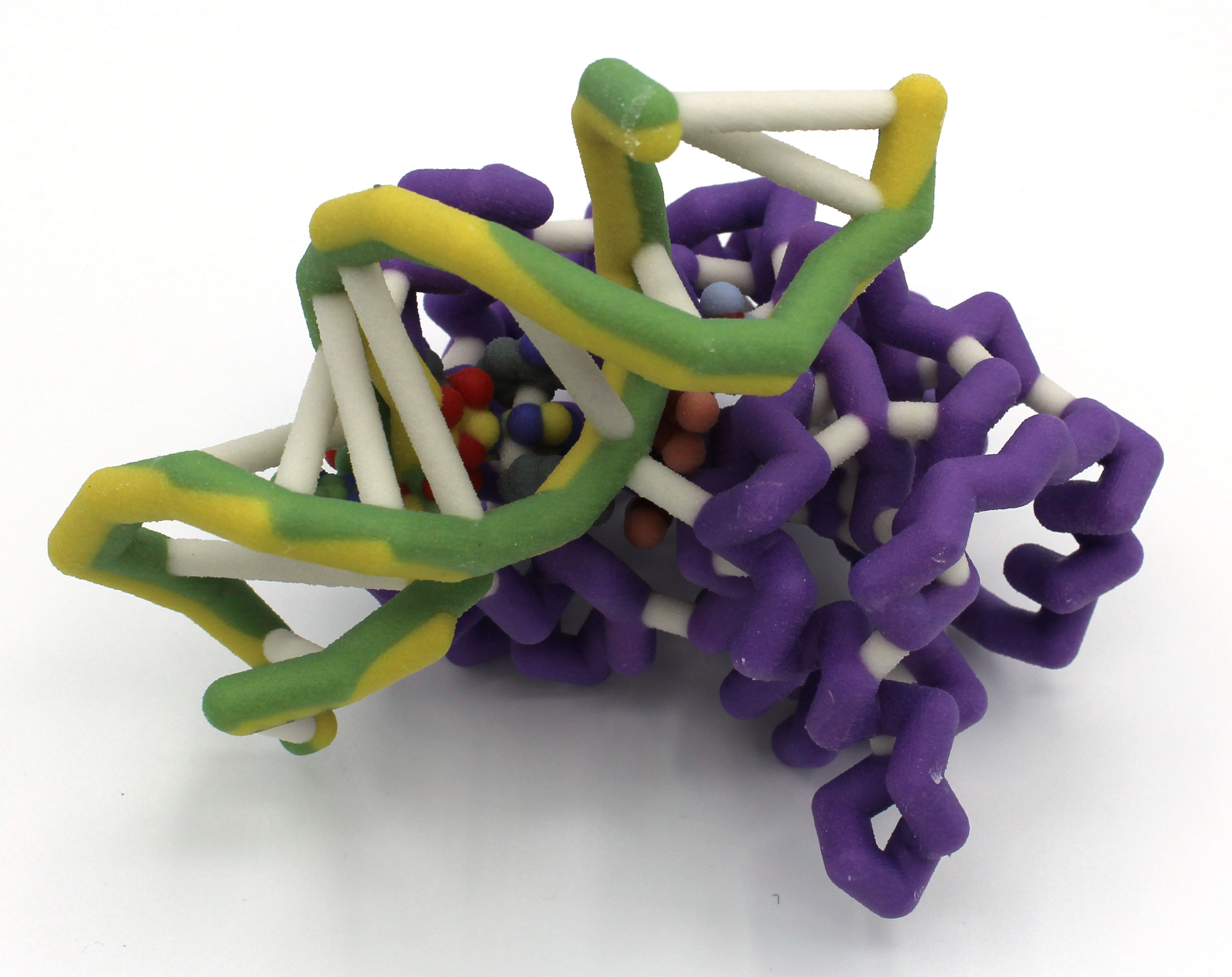
PDB files: 4OX9.pdb, 1J5E.pdb
Primary Citations: 4ox9: Dunkle, J. A., Vinal, K., Desai, P. M., Zelinskaya, N., Savic, M., West, D. M., . . . Dunham, C. M. (2014). Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proceedings of the National Academy of Sciences, 111(17), 6275-6280.
1j5e: Wimberly, B. T., Brodersen, D. E., Clemens, W. M., Morgan-Warren, R. J., Carter, A. P., Vonhrein, C., . . . Ramakrishnan, V. (2000). Structure of the 30S ribosomal subunit. Nature. 407(6802), 327-39.
Abstract: Aminoglycosides are broad-spectrum antibiotics common in clinical, veterinary, and agricultural settings and are often reserved for treating severe bacterial infections. With antibiotic resistance becoming a global crisis, understanding the mechanisms by which bacteria attain such resistance is more urgent than ever. One such mechanism specific to aminoglycosides is ribosomal modification by methyltransferases (RMTases). RMTases pose a significant threat as they grant simultaneous resistance to various aminoglycosides and are transmitted between species through genetic exchanges.  The protein NpmA is a RMTase that confers blanket resistance to aminoglycosides by transferring a methyl group to the A1408 nucleotide in helix 44 of the 30S ribosomal subunit. Normally, aminoglycosides alter decoding centers of ribosomes, leading to inaccuracies in protein production and triggering cell death. Methylation of the nucleotide A1408, however, makes helix 44 unrecognizable to aminoglycosides. This small chemical change has a massive effect rendering an entire class of antibiotics ineffective. To better promote understanding of this resistance mechanism, we designed a new three-dimensional model to demonstrate the interactions between NpmA and helix 44. We took special care to highlight tryptophan residues 107 (W107) and 197 (W197), which together function on pi stacking to stabilize A1408 once it is flipped, as well as arginine 207 (R207) and the residue responsible for positioning R207, glutamate 146 (E146). Arginine 207 is static in both the bound and free forms of NpmA but would sterically clash with the phosphate group of A1408 in the absence of base flipping. Upon binding of NpmA to rRNA, a conformational change brings E146 into position to form a hydrogen bond with R207. The hydrogen bond acts as a buttress, forcing A1408 to flip to avoid a steric clash. Further research is necessary to elucidate the mechanisms behind NpmA as well as to counteract its activity in aminoglycoside-resistant bacteria.
The protein NpmA is a RMTase that confers blanket resistance to aminoglycosides by transferring a methyl group to the A1408 nucleotide in helix 44 of the 30S ribosomal subunit. Normally, aminoglycosides alter decoding centers of ribosomes, leading to inaccuracies in protein production and triggering cell death. Methylation of the nucleotide A1408, however, makes helix 44 unrecognizable to aminoglycosides. This small chemical change has a massive effect rendering an entire class of antibiotics ineffective. To better promote understanding of this resistance mechanism, we designed a new three-dimensional model to demonstrate the interactions between NpmA and helix 44. We took special care to highlight tryptophan residues 107 (W107) and 197 (W197), which together function on pi stacking to stabilize A1408 once it is flipped, as well as arginine 207 (R207) and the residue responsible for positioning R207, glutamate 146 (E146). Arginine 207 is static in both the bound and free forms of NpmA but would sterically clash with the phosphate group of A1408 in the absence of base flipping. Upon binding of NpmA to rRNA, a conformational change brings E146 into position to form a hydrogen bond with R207. The hydrogen bond acts as a buttress, forcing A1408 to flip to avoid a steric clash. Further research is necessary to elucidate the mechanisms behind NpmA as well as to counteract its activity in aminoglycoside-resistant bacteria.
Lane College
Ryan Billings, Giovanni Avila, Alexis Craft, Keviyanah Gilmore and Dajah Patterson

Faculty Advisors: Candace Jones, Ph.D. and Melanie Van Stry, Ph.D.
Mycobacterium tuberculosis MazF-mt6 toxin protein
PDB file: 5UCT.pdb
Primary Citation: Hoffer EA, Miles SJ and Dunham CM. (2017). The structure and function of Mycobacterium tuberculosis MazF-mt6 provides insights into conserved features of MazF endonucleases. Journal of Biological Chemistry 292(19):7718-26.
Abstract: Toxin-antitoxin systems are present in bacterial and archaeal genomes and regulate cellular survival in response to stressors including antibiotics and host immune responses. The toxin-antitoxin systems all consist of a toxin protein that blocks essential cellular functions such as translation, and either an antitoxin RNA or protein that inhibits toxin function through a variety of mechanisms. Escherichia coli has at least 36 assumed toxin-antitoxin gene pairs, whereas the pathogenic Myocobacterium tuberculosis has over 90 toxin-antitoxin pairs. The E. coli MazF toxin proteins are ribonucleases that cleave cellular mRNA once stress has been encountered, while the nine M. tuberculosis MazF family cleave mRNA, tRNA, or rRNA. MazF-mt6 from M. tuberculosis cleaves helix 70 of the 23S rRNA. Here, we have designed a 3D of model of the MazF-mt6 protein using Jmol that highlights the  overall architecture and the active site based on the crystal structure solved by Dr. Dunham and colleagues (PDB 5UCT; Hoffer et al. 2017 JBC 292(19):7718-26). MazF-mt6 protein has Pemk-like fold with three alpha-helices, shown in grey, and 2 beta-sheets shown in purple, one of which folds into a five-stranded beta-barrel fold similar to the SH3 domain. The active site is solvent accessible and contains several charged or polar residues that may be involved in catalysis. Our 3D model highlights the critical residues in the active site. Site-directed mutagenesis studies reveal that Asp10, Arg13, Lys11, and Thr36 are necessary for MazF-mt6 induced growth arrest, whereas Thr37 and Thr38 are not. In addition, Asp10, Arg13, and Thr36 are necessary for enzymatic activity, suggesting that they are part of a catalytic triad. Designing 3D protein models enables students to explore complex molecular structure-function relationships.
overall architecture and the active site based on the crystal structure solved by Dr. Dunham and colleagues (PDB 5UCT; Hoffer et al. 2017 JBC 292(19):7718-26). MazF-mt6 protein has Pemk-like fold with three alpha-helices, shown in grey, and 2 beta-sheets shown in purple, one of which folds into a five-stranded beta-barrel fold similar to the SH3 domain. The active site is solvent accessible and contains several charged or polar residues that may be involved in catalysis. Our 3D model highlights the critical residues in the active site. Site-directed mutagenesis studies reveal that Asp10, Arg13, Lys11, and Thr36 are necessary for MazF-mt6 induced growth arrest, whereas Thr37 and Thr38 are not. In addition, Asp10, Arg13, and Thr36 are necessary for enzymatic activity, suggesting that they are part of a catalytic triad. Designing 3D protein models enables students to explore complex molecular structure-function relationships.
Milwaukee School of Engineering
Alexandra Berkowicz, Bella Cebrera Brito, Muskan Kanungo, Sabrina Mierswa, Krishna Persaud, Ben Pung and Guinevere Sieradzki
Faculty Advisor: Anne Alexander, Ph.D.
I Can Barely Hold On: Analyzing the HigB/HigA Toxin/Antitoxin Interaction in the Stringent Response
PDB file: 4MCT.pdb
Primary Citation: Schureck, M., Dunkle, J., Maehigashi, T., & Dunham, C. (2015). Structure of P. vulgaris HigB toxin. Journal of Biological Chemistry. doi:10.2210/pdb4px8/pdb
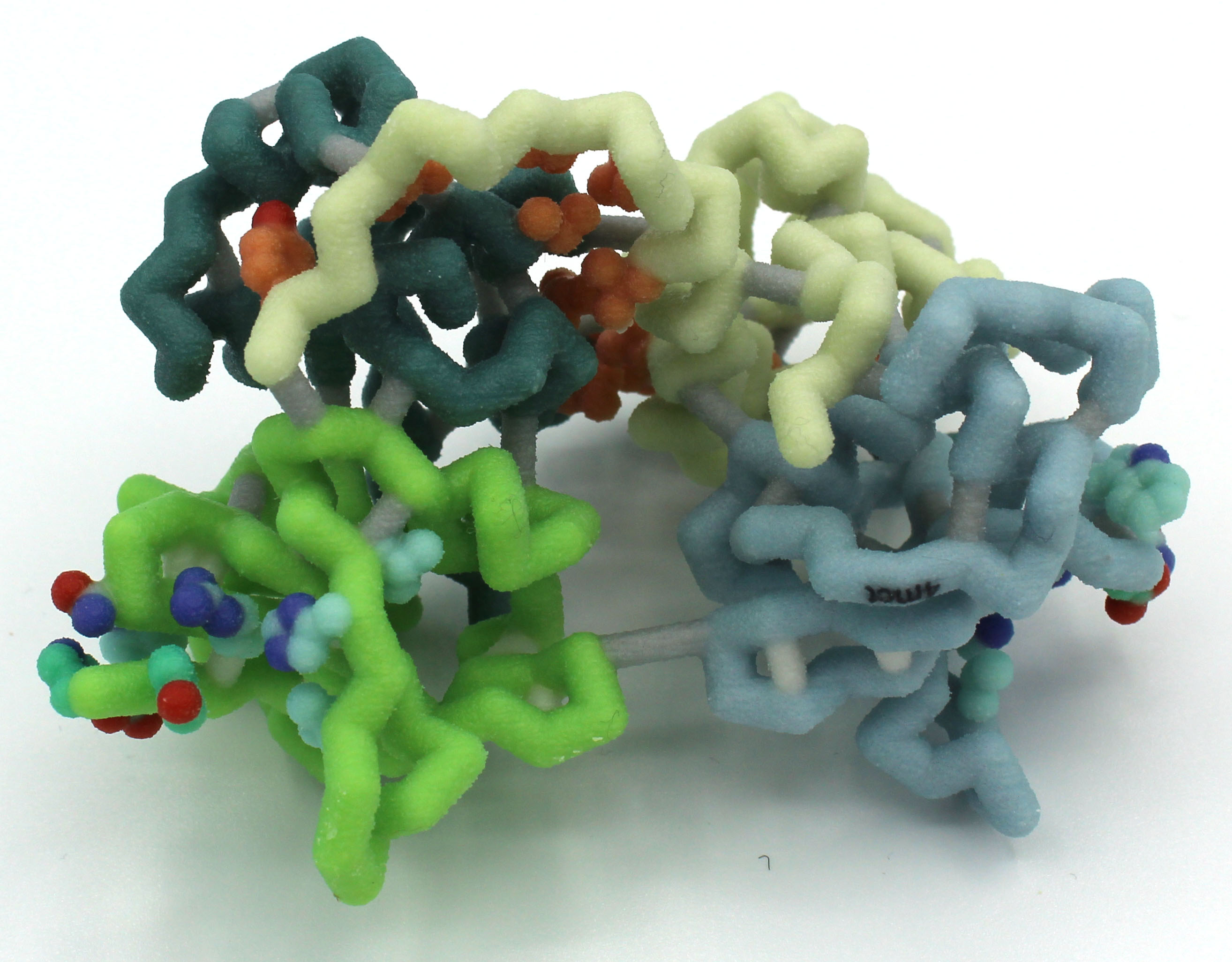
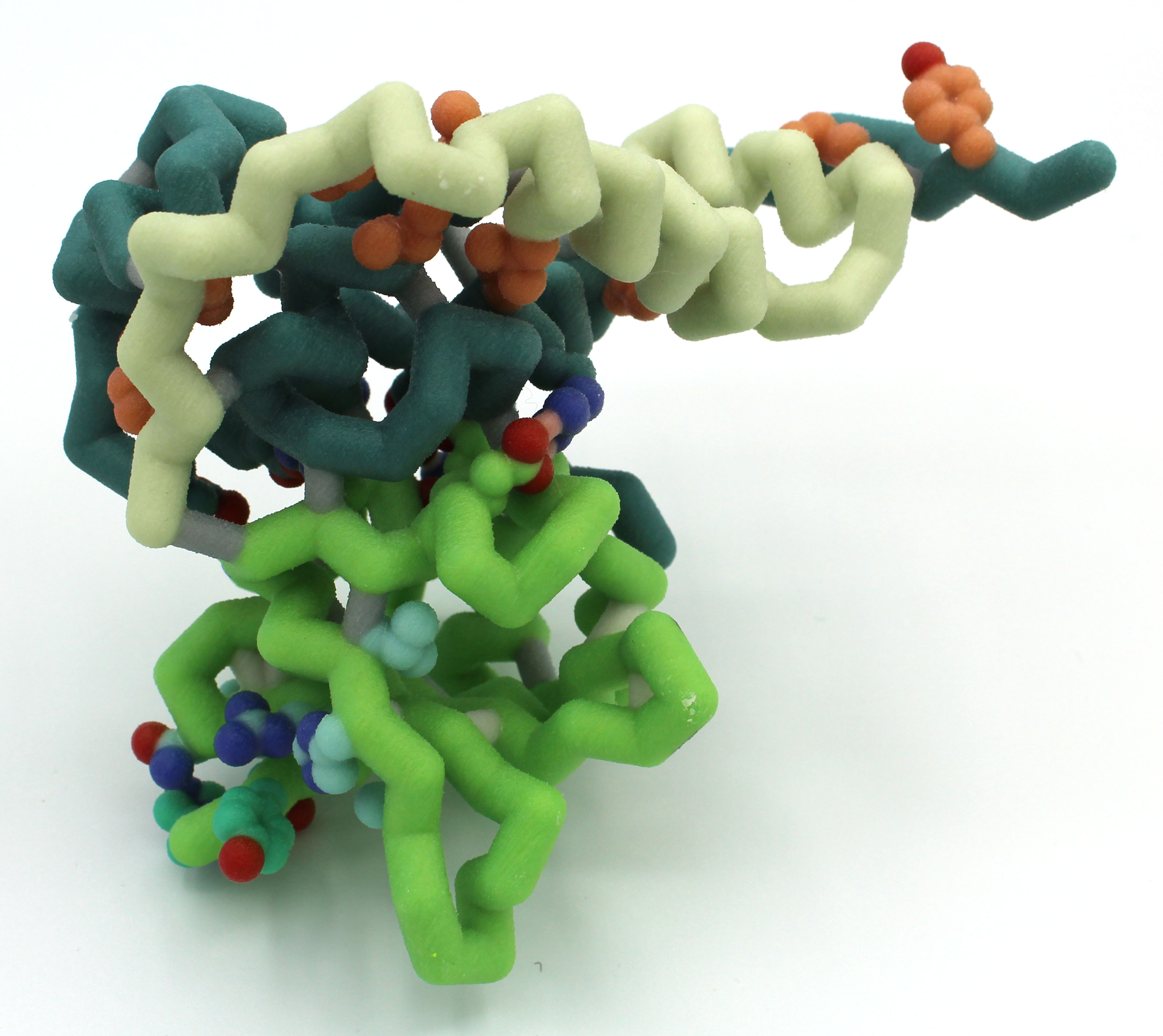
Abstract: Society is losing its ability to combat serious bacterial infections as bacteria develop more mechanisms of antibiotic resistance. One of these mechanisms is the stringent response, which is triggered by stressful conditions like an exposure to antibiotics. The bacteria enter a state of dormancy where major functions like replication and protein synthesis are slowed until conditions improve. The stringent response is often mediated by proteins in the RelE family that can include toxin-antitoxin complexes. HigB toxin, a member of the RelE family due to its adjacent alpha helix and antiparallel beta sheet, degrades mRNA before it can be translated into protein by the ribosome. This reduces the energy used for protein synthesis, allowing the cell to enter a state of dormancy.

In the absence of stressful conditions, HigB is present in the cells but is bound to the antitoxin HigA. The interactions between HigB and HigA sterically inhibit the activity of HigB, thus allowing translation. This is unlike other RelE antitoxins because HigA does not directly interfere with the active site on HigB. Instead, these steric effects prevent HigB from reaching the mRNA in the ribosome’s E site and cleaving it. The antitoxin HigA forms a dimer through hydrophobic interactions at their C-terminal tails. Tetramer development occurs when the antitoxin dimer interacts with two toxin molecules. The stringent response triggers the degradation of the HigA dimer, which allows the HigB toxins to cleave mRNA.
Our model highlights the active site on HigB responsible for mRNA attachment and degradation. This active site comprises residues 53, 54, 56, and 71 – 73. Nearby, a hook made of residues 90 – 92 pulls the mRNA into the active site. Our model also highlights how HigA and HigB interact through a hydrogen bonding network, hydrophobic interactions, and ionic interactions.
Nova Southeastern University
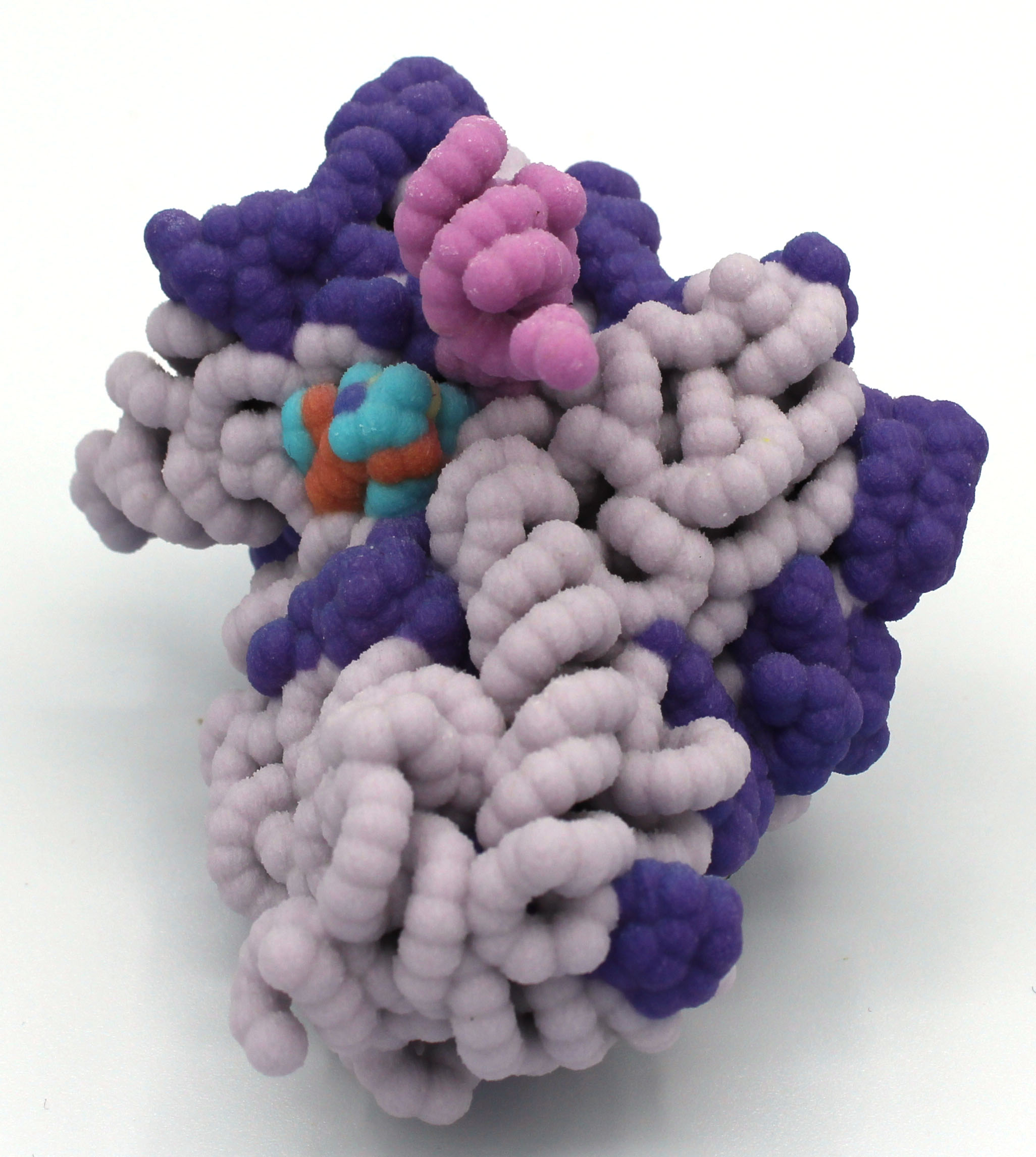
Carolina Alzamora, Alesa Chabbra, Helana Ghali, Marina Handal, Mahima Kathiria, Alexander Martinek, Jenny Nguyen, Chandni Patel, Shreja Patel, Antoine Pham
Faculty Advisor: Emily Schmitt Lavin, Ph.D.
Mechanism of Endonuclease Cleavage by the HigB Toxin
PDB file: 4ZSN.png
Primary Citation: Marc A. Schureck, Adrienne Repack, Stacey J. Miles, Jhomar Marquez, Christine M. Dunham; Mechanism of endonuclease cleavage by the HigB toxin, Nucleic Acids Research, Volume 44, Issue 16, 19 September 2016, Pages 7944–7953, https://doi.org/10.1093/nar/gkw598

Abstract: The proteins, toxin HigB and antitoxin HigA, are part of a type II toxin-antitoxin system that cleaves ribosome-bound mRNAs in bacteria. Chemical or physical stresses, such as extreme temperatures and lack of nutrients, lead to antitoxin degradation that releases HigB. Temporary inhibition of growth by HigB allows cells to more efficiently utilize current resources. Although HigB is referred to as a toxin, it is not inherently detrimental and can be beneficial to cellular survival. Bacteria that use this mechanism include Escherichia coli, Pseudomonas aeruginosa, and Proteus vulgaris. As such, HigB is of clinical interest due to its contribution in prolonging bacterial infections through antibiotic tolerance (persistence). When cells are not in a stressed state, HigA binds to and inactivates HigB, allowing growth and translation to proceed. Details of HigB in complex with the ribosome as well as residues important for mRNA cleavage were found in the Protein Data Bank file, 4ZSN, and imported into Jmol, a protein visualization software. The 4ZSN file consisted of several nucleic acid and protein bundles. Bundle 1 (small ribosomal subunit) was examined in more detail.
University of Minnesota Rochester
Heather Berg, Joshua Dennis, Tabitha Guenther, Denika Kerska and Tucker Rients
Faculty Advisor: Cassidy Terrell, Ph.D.
A Toxic Tragedy: The Molecular Story of the Toxin YafQ’s Regulation of Protein Translation
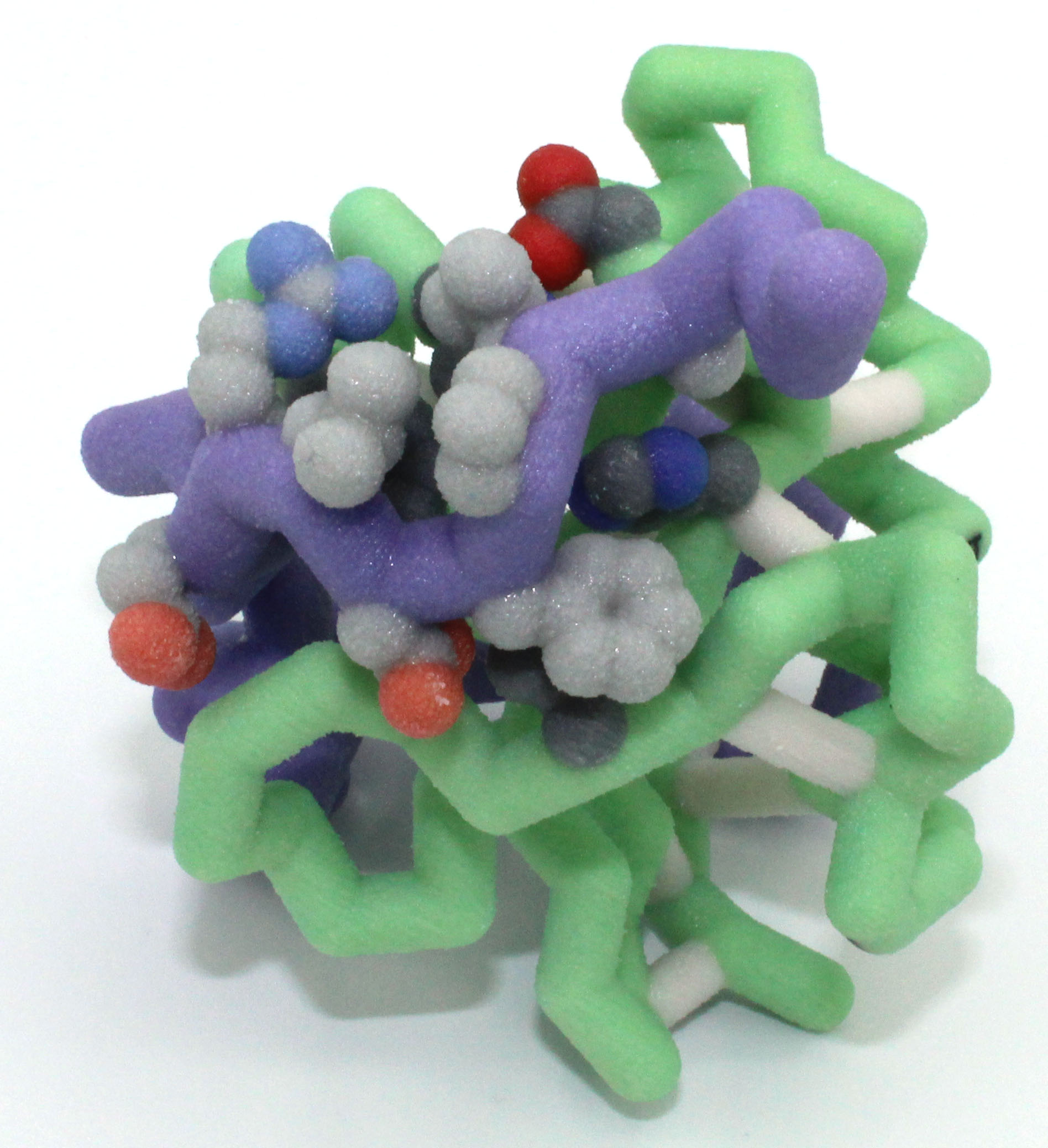
PDB file: 4Q2U.pdb
Primary Citation: Maehigashi, T., Ruangprasert, A., Miles, S. J., & Dunham, C. M. (2015). Molecular basis of ribosome recognition and mRNA hydrolysis by the E. coli YafQ toxin. Nucleic Acids Research, 10.1093/nar/gkv791.
Abstract: During times of cellular stress, Escherichia coli uses YafQ (Chain B of PDB ID: 4Q2U), an mRNA interferase toxin, to prevent protein translation. For example, when the local food supply for the bacterium is low this triggers a stress response so that the bacterium halts translation, effectively avoiding unnecessary use of cellular resources. Here, the cell activates the toxin by degrading the bound antitoxin protein (DinJ) (Chain A of PDB ID: 4Q2U). After dissociation of DinJ, YafQ is able to bind to and catalyze the hydrolysis of mRNA that is concurrently bound to the ribosome. This effectively degrades the mRNA strand and halts translation. Based on this information, we designed a three-dimensional physical model demonstrating how DinJ binding to YafQ effectively blocks mRNA binding. In this model, key binding residues on DinJ  (Ala46, Leu47, Pro48, Phe49, Asp50, Leu51, Arg52 and Glu53) are rendered in spacefill showing how completely this antitoxin protein blocks the mRNA binding site of YafQ. The blocked active site residues of YafQ (His50, His63, Asp67, His87 and Phe91) are rendered in spacefill and CPK to demonstrate non-covalent interactions with DinJ. These are the same residues that interact with the mRNA. Additionally, the physical model of the antitoxin DinJ is truncated, where residues 3-42 are not shown for clarity in the model because they do not interact with the toxin YafQ. Understanding the molecular level interactions between the toxin and its binding partners could lead to the development of antibiotics that target YafQ and halt protein translation in bacteria.
(Ala46, Leu47, Pro48, Phe49, Asp50, Leu51, Arg52 and Glu53) are rendered in spacefill showing how completely this antitoxin protein blocks the mRNA binding site of YafQ. The blocked active site residues of YafQ (His50, His63, Asp67, His87 and Phe91) are rendered in spacefill and CPK to demonstrate non-covalent interactions with DinJ. These are the same residues that interact with the mRNA. Additionally, the physical model of the antitoxin DinJ is truncated, where residues 3-42 are not shown for clarity in the model because they do not interact with the toxin YafQ. Understanding the molecular level interactions between the toxin and its binding partners could lead to the development of antibiotics that target YafQ and halt protein translation in bacteria.
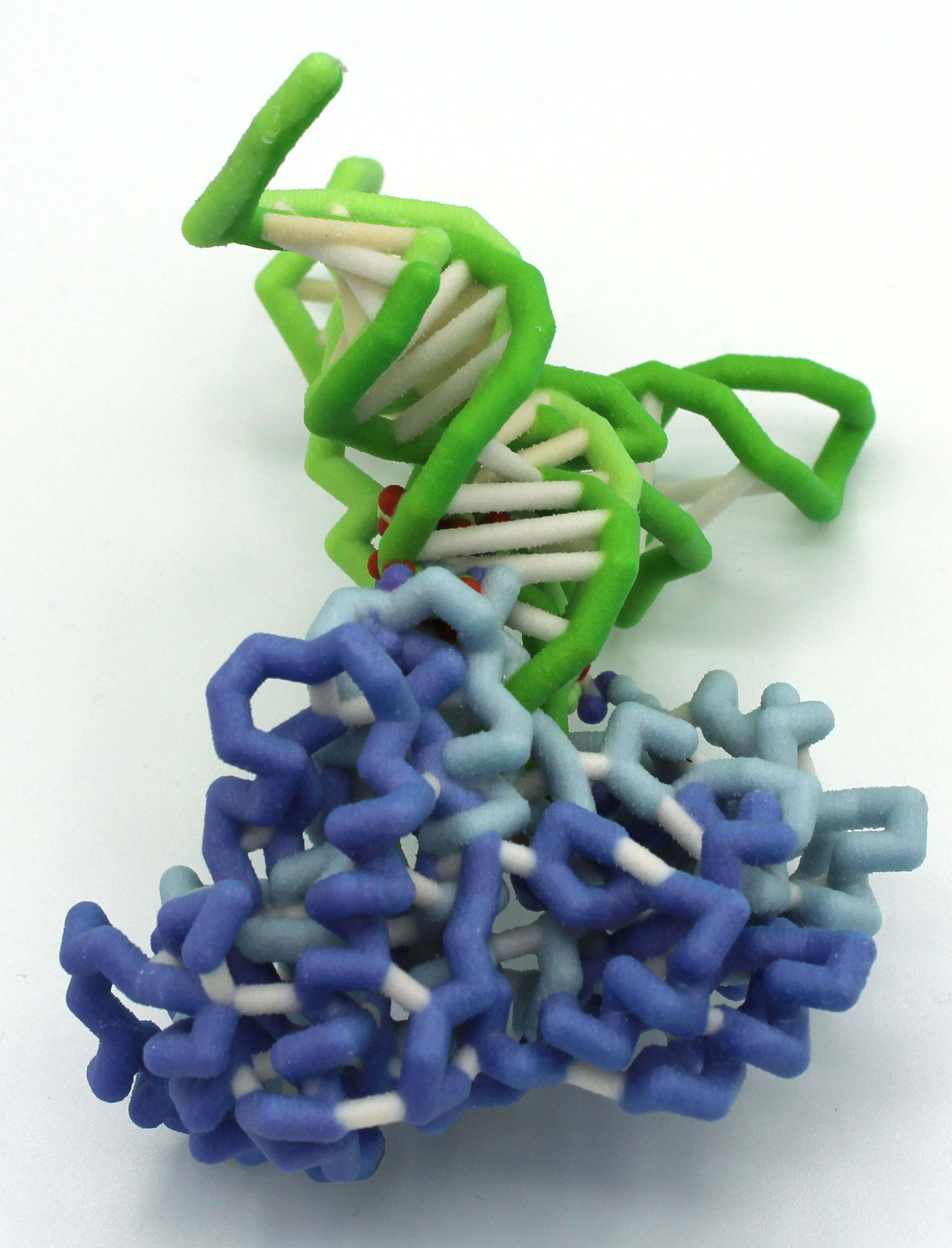
University of San Diego
Sharon Shania and Natalie Botros, Nicholas Lee, Kalii Faustino, Ireland Vandentop, Hanna Blythe, Ryan McCool, Tatiana Mancham, Amelia Smith, Tabitha Wilbur
Faculty Advisor: Ellis Bell, Ph.D.
uL1 and E-tRNA Complex
PDB files: 5uq7 and 5uq8
Primary Citation: Zhang, et al., (2018). Alternative Mode of E-Site tRNA Binding in the Presence of a Downstream mRNA Stem Loop at the Entrance Channel. Structure 26, 437–445.
Abstract: Ribosomes play an integral role in protein synthesis. Within the crystallized 70S ribosome complex (PDB: 5UQ7), there are 50 proteins and 6 different RNA species (1). To further understand the structure and function of the proteins within this complex, we modeled Thermus thermophilus ribosomal protein L1 (uL1) and its interaction with E (exit)-site tRNA (1). The L1 stalk of the 50S subunit is a conserved and highly dynamic structure made of protein L1 (uL1) and RNA binding site (3). uL1 is of interest due to its observed direct interaction with E-tRNA where it appears to block the path of the tRNA for its exit from the ribosome (2,3). Since the exit path is blocked by uL1, both E-tRNA and uL1 undergo an open (uL1out/EouttRNA) to  closed (uL1in/EintRNA) conformational change involved in the release of tRNA (1,2). Though conformational changes were observed, the specific residues interacting between uL1 and E-tRNA have not been further studied. We examined H-bonds and polar interactions between uL1 and E-tRNA that may play a role in the regulation of this conformational change and are illustrated in the model. We propose four interactions between uL1 and tRNA involving: R52 to C61, R53 to C62, R129 to C56, and P133 to C56, where atomic distances appear similar between open and closed conformations for all interactions except the R129 and C56 interaction. In closed conformation (PDB: 5UQ7), R129 and C56 are 2.63 Å apart and in an open conformation (PDB: 5UQ8), R129 and C56 are 5.15 Å apart. The difference in atomic distance between R129 and C56 in each conformation may indicate the importance of this interaction in regulating the structural changes observed. As observed, EouttRNA directly impacts uL1 conformation.
closed (uL1in/EintRNA) conformational change involved in the release of tRNA (1,2). Though conformational changes were observed, the specific residues interacting between uL1 and E-tRNA have not been further studied. We examined H-bonds and polar interactions between uL1 and E-tRNA that may play a role in the regulation of this conformational change and are illustrated in the model. We propose four interactions between uL1 and tRNA involving: R52 to C61, R53 to C62, R129 to C56, and P133 to C56, where atomic distances appear similar between open and closed conformations for all interactions except the R129 and C56 interaction. In closed conformation (PDB: 5UQ7), R129 and C56 are 2.63 Å apart and in an open conformation (PDB: 5UQ8), R129 and C56 are 5.15 Å apart. The difference in atomic distance between R129 and C56 in each conformation may indicate the importance of this interaction in regulating the structural changes observed. As observed, EouttRNA directly impacts uL1 conformation.
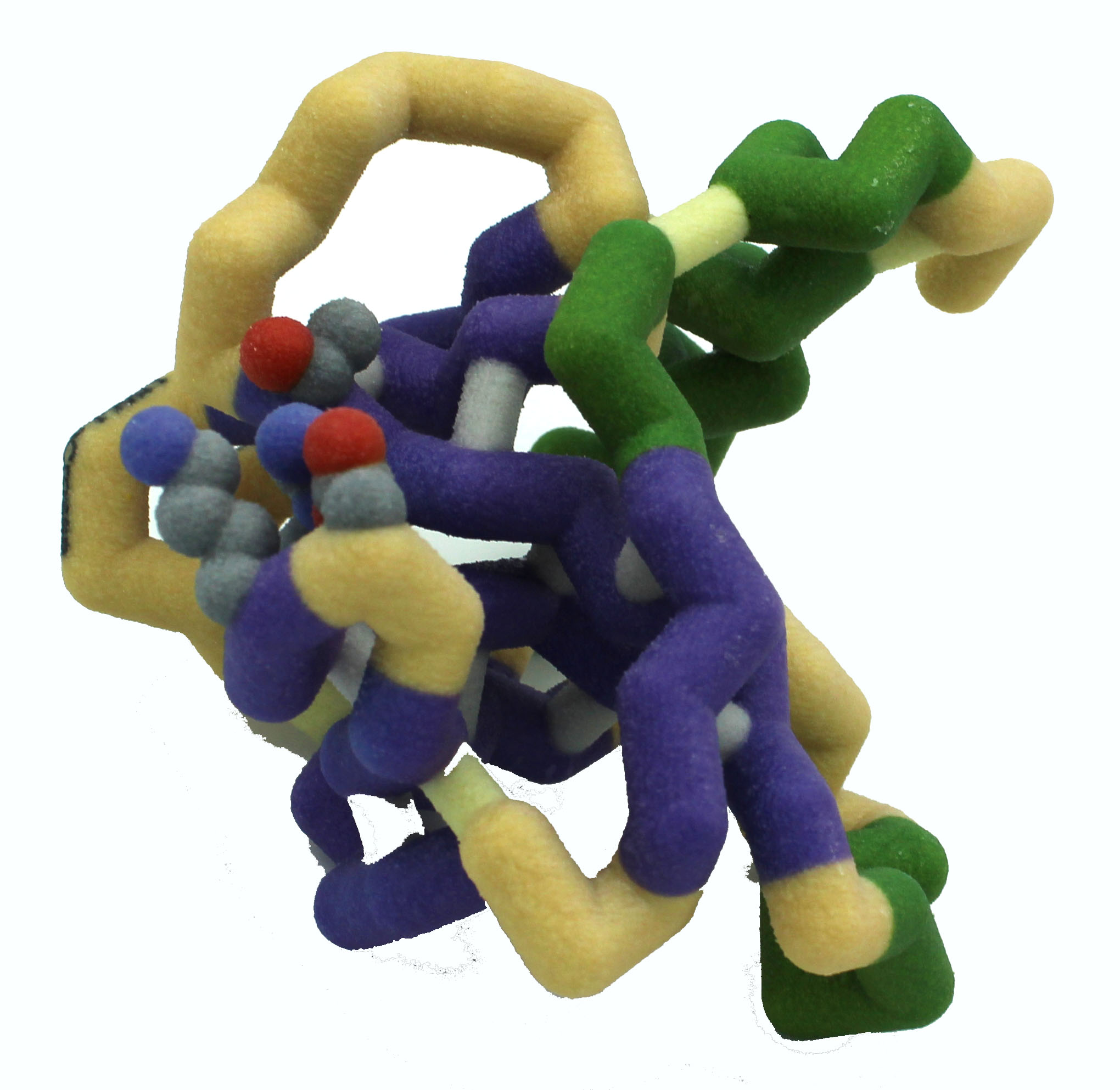
University of Wisconsin La Crosse
JB Becker, Gina Wade
Faculty Advisor: Basudeb Bhattacharyya, Ph.D.
MazF-mt6 and Large Ribosomal Subunit of Mycobacterium tuberculosis
PDB files: 5UCT.pdb (toxin/antitoxin); 5V7Q.pdb (ribosome)
Primary Citations: 5UCT: Hoffer, E. D., Miles, S. J., Dunham C. M. (2017). The structure and function of Mycobacterium tuberculosis MazF-mt6 toxin provide insights into conserved features of MazF endonucleases. J Biol Chem. 292(19), 7718-7726.
5V7Q: Yang, K., Chang, J.Y., Cui, Z., Li, X., Meng, R., Duan, L., Thongchol, J., Jakana, J., Huwe, C.M., Sacchettini, J.C., and Zhang, J. (2017). Structural insights into species-specific features of the ribosome from the human pathogen Mycobacterium tuberculosis. Nucleic Acids Res. 45, 10884-10894.

Abstract: Bacteria are constantly surrounded by threats much larger than themselves. When faced with host immune defenses or antibiotic treatments, such vulnerable organisms need to defend themselves, often at the expense of human health. Toxin-antitoxin systems are one of these defenses and are utilized during periods of stress. Mycobacterium tuberculosis, the causative agent of tuberculosis, utilizes the dimer toxin MazF-mt6 to cleave its own ribosome at helix 70 between the large (50s) and small (30s) ribosomal subunits, thus preventing protein synthesis. Residues Asp-10, Lys-11, Arg-13, and Thr-36 are important in this catalytic cleavage and form the active site of MazF-mt6. The carbonyl of Asp-10 forms electrostatic interactions with the catalytic Arg-13 while Thr-36 and Lys-11 stabilize the transition state of the helix 70 RNA to be cleaved. Once a dormant state is achieved, the bacteria waits until the threat has passed before expressing its antitoxin, MazE-mt6, to inactivate MazF-mt6 and prevent further ribosome cleavage. MazE-mt6 binding disrupts the catalytic Arg-13 to inactivate the toxin. Proteases degrade the antitoxin in response to another period of stress, freeing MazF-mt6. Variants of MazF-mt6 and non-MazF toxin systems are found in other human pathogens as well, such as Escherichia coli and Bacillus subtilis. Using the MazF-mt6 structure as a model system, research on such ubiquitous bacterial defense mechanisms opens a promising path to developments in novel antibacterial treatments for human-targeting pathogens.
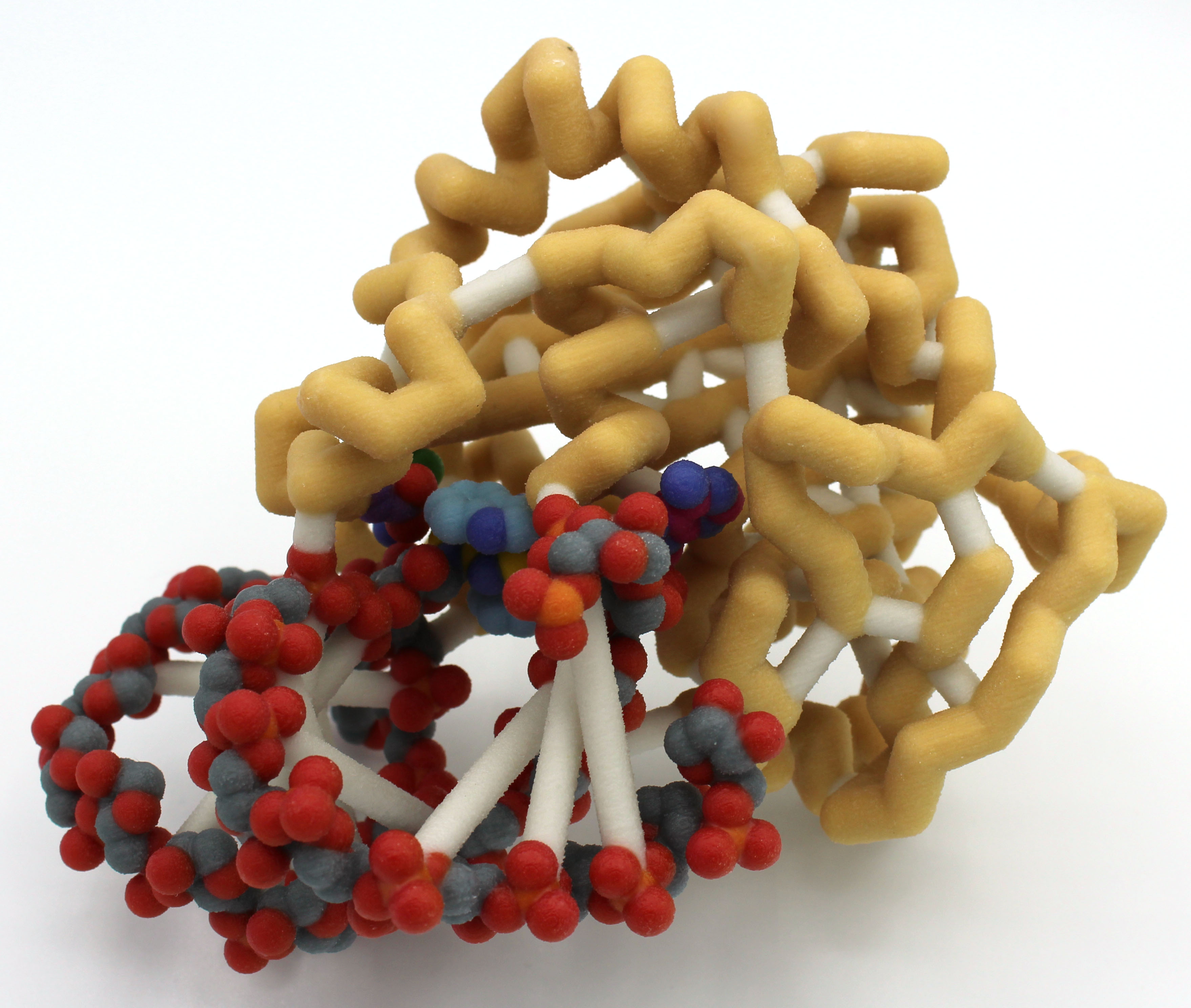
University of Wisconsin La Crosse
Julian Grosskopf, Paige Goethel, Bryton Foate and Rachel Senft
Faculty Advisor: Basudeb Bhattacharyya, Ph.D.
Aminoglycoside-Resistance Methyltransferase NpmA
PDB file: 4OX9.pdb
Primary Citation: Dunkle, J. A., et al. “Molecular Recognition and Modification of the 30S Ribosome by the Aminoglycoside-Resistance Methyltransferase NpmA.” Proceedings of the National Academy of Sciences, vol. 111, no. 17, 2014, pp. 6275–6280., doi:10.1073/pnas.1402789111.

Abstract: The novel plasmid-mediated aminoglycoside-resistance rRNA methyltransferase A (NpmA) is used to confer bacterial resistance to aminoglycosides (antibiotics) that target the 30S ribosomal subunit. With the increasing number of antibiotic-resistant infectious strains, the study of antibiotic-resistant mechanisms becomes a necessity for the push against these organisms. NpmA recognizes adenosine 1408 (A1408), a member of the 30S subunit rRNA, and alters this site to act in masking it from aminoglycosides. Because NpmA modifies the aminoglycoside drug target, and not the drug itself, it confers class-wide resistance against these drugs. NpmA does this specifically by methylation at A1408, producing m1A1408 with the help of an S-adenosyl-L-methionine (SAM), bound in the class I methyltransferase SAM-binding fold. Conformational changes within NpmA after the binding of the SAM molecule allow the conformational “flipping” of A1408 away from its RNA double-helical structure and into the NpmA catalytic site. The orchestration of this event brings many critical residues into symphony with one another, in most cases showing that a single missing residue compromises the catalytic activity of NpmA. The rRNA structure recognized by NpmA is heavily conserved, thus offering NpmA as a great model system to begin work with inhibitors targeting bacterial resistance mechanisms as unique as this.
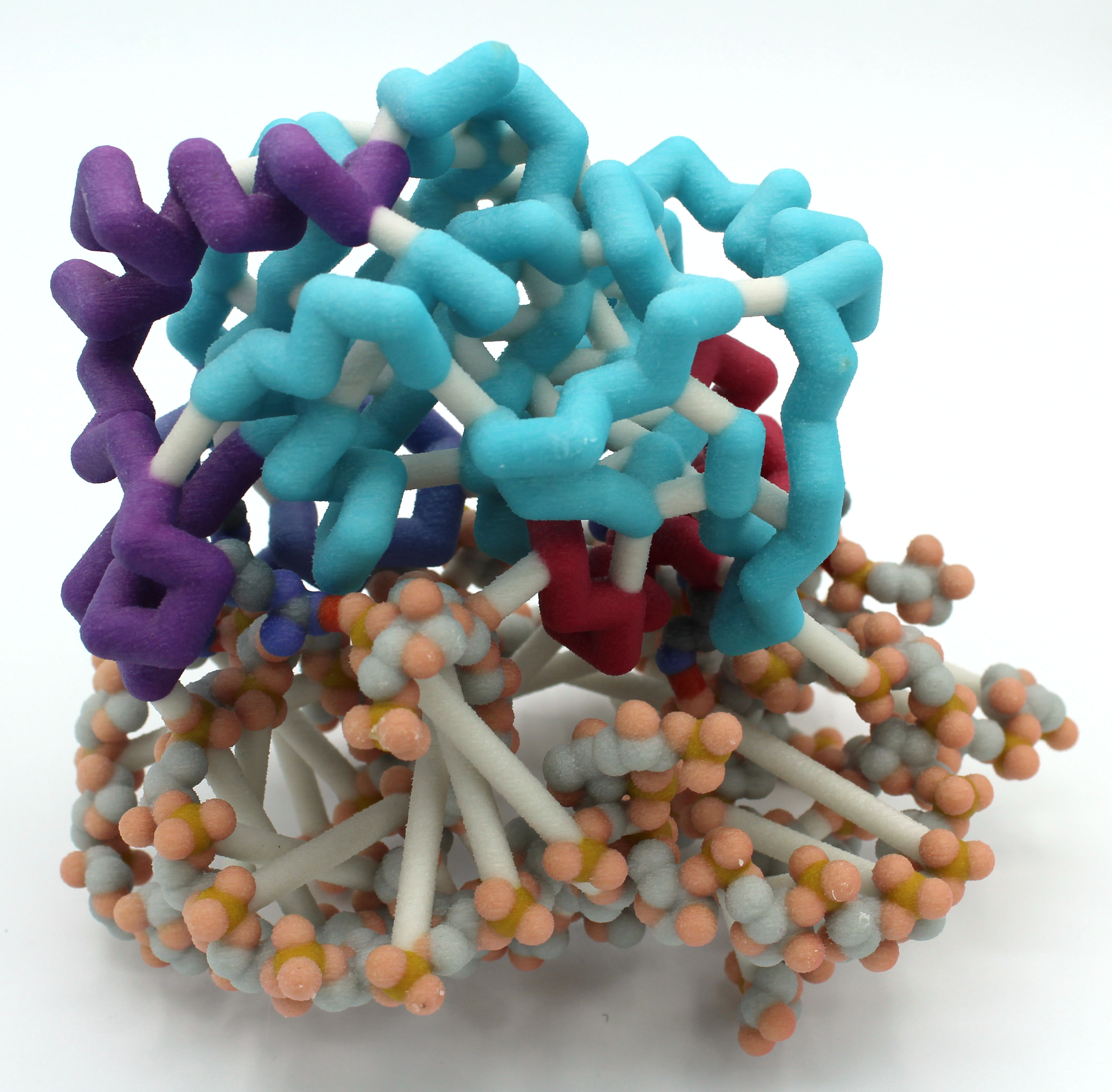
University of Wisconsin La Crosse
Ross Soens, Carter Caya and Gaoying Vue
Faculty Advisor: Basudeb Bhattacharyya, Ph.D.
Ross Soens, Carter Caya and Gaoying Vue
PDB file: 4OX9.pdb
Primary Citation: Dunkle, Jack & Vinal, Kellie & M Desai, Pooja & Zelinskaya, Natalia & Savic, Miloje & West, Dayne & Conn, Graeme & Dunham, Christine. (2014). Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proceedings of the National Academy of Sciences of the United States of America. 111. 10.1073/pnas.1402789111.
Abstract: Roughly 33% of E. coli cases in the United States are resistant to one or more commonly used antibiotics. One of the culprits of this trend in antibiotic resistance is aminoglycoside-resistance. Aminoglycosides are a class of antibiotics that target the highly-conserved sequence and structure of bacterial ribosomes thus interfering with necessary protein production. The plasmid-mediated aminoglycoside-resistance rRNA methyltransferase A (NpmA) in E. coli confers resistance to aminoglycoside antibiotics. NpmA modifies the drug binding site on the ribosome by binding specifically to the 30S ribosomal subunit helices h24, h27, h44, and h45 using linkers between beta-strands 2/3, 5/6, and 6/7. Once bound, NpmA methylates adenine 1408 on helix 44 of the 30S subunit using a S-adenosylmethionine (SAM) molecule. This methylation of the 30S subunit provides resistance to common aminoglycosides such as kanamycin. A three-dimensional model of NpmA has been assembled to exhibit its 30S ribosomal subunit specific binding site. With this model, we can help visualize this specific binding of NpmA with the hopes of generating novel inhibitors to combat aminoglycoside resistance.
University of Wisconsin Stevens Point
Langenfeld, Noah J., Freese, Kaitlin K., Harper, Logan E., Schuetze, Samantha C. and Schultz, Tehya D.
Faculty Advisor: Amanda Jonnson, Ph.D.
Bacterial Aminoglycoside Resistance via NpmA rRNA Methyltransferase
PDB file: 4OX9.pdb
Primary Citation: Dunkle JA, Vinal K, Desai PM, Zelinskaya N, Savic M, West DM, Conn GL, Dunham CM. 2014. Molecular recognition and modification of the 30S ribosome by the aminoglycoside-resistance methyltransferase NpmA. Proc. Natl. Acad. Sci.111: 6275-6280.
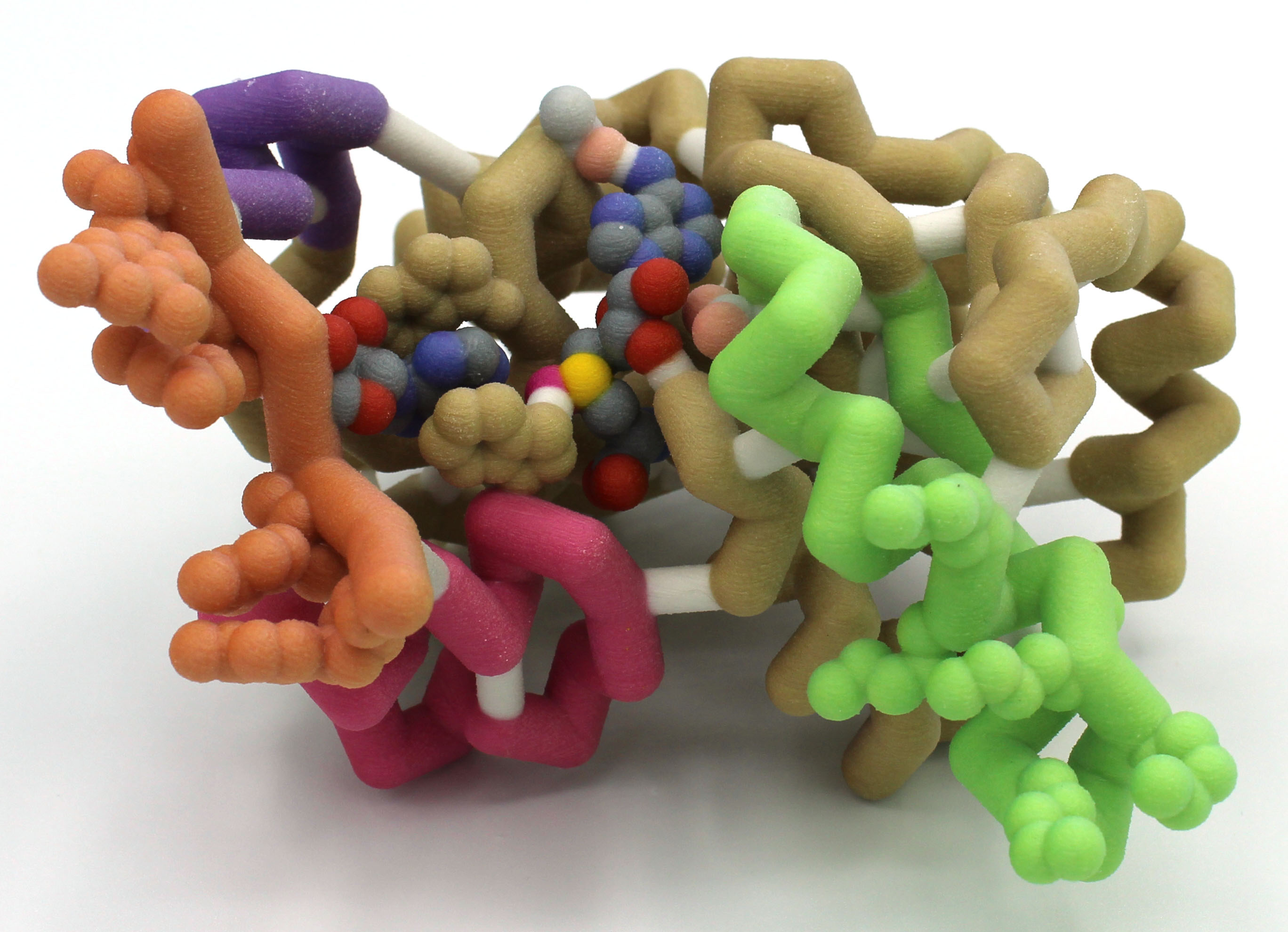
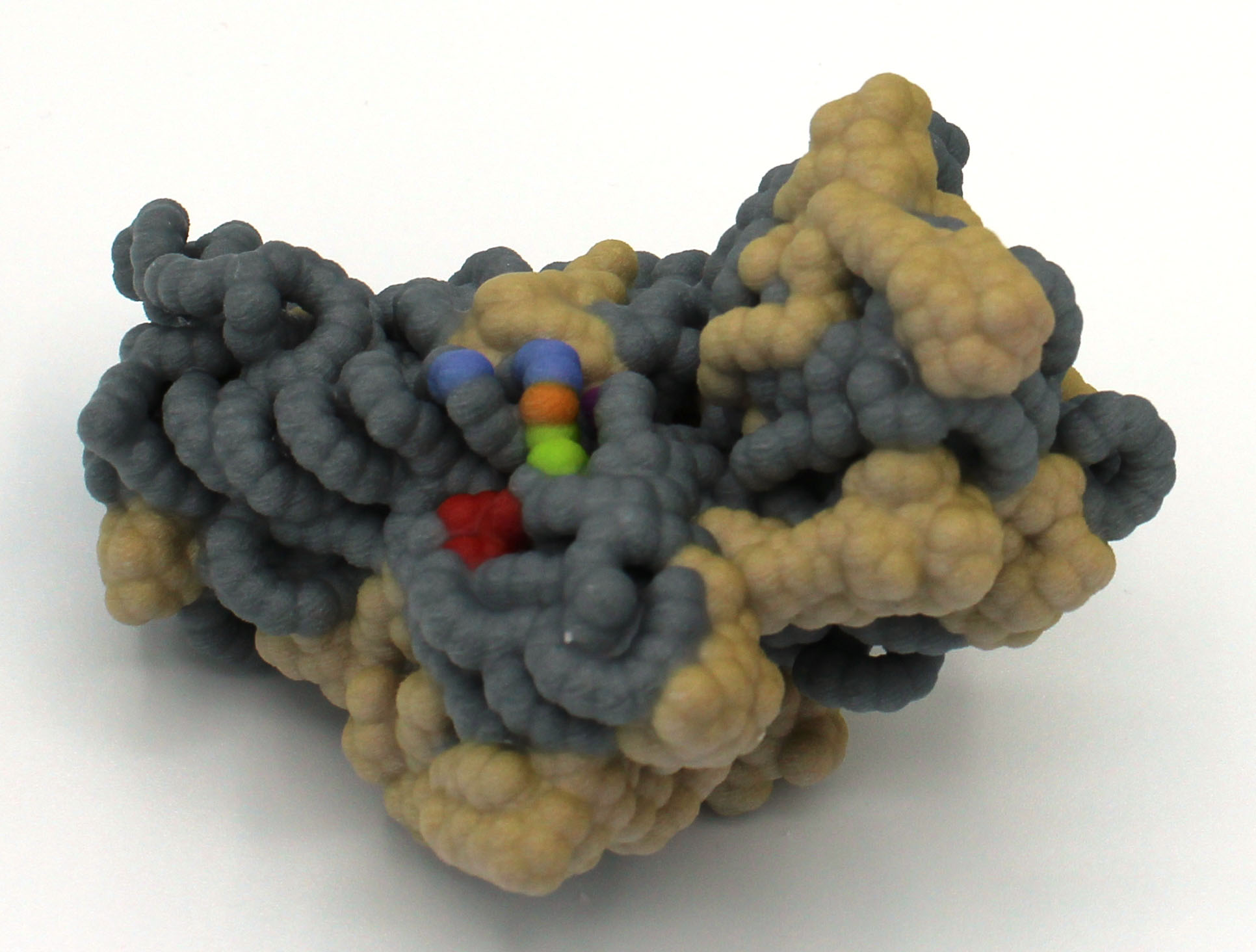
Abstract: The use of antibiotics in the medical community has led to cures for many common bacterial infections. However, due to their short life cycles, bacteria have evolved several mechanisms to combat antibiotics and inhibit their effects. Aminoglycosides are a broad class of antibiotics that effect proofreading mechanisms in the translation of mRNA and cause bacteria to produce non-functional proteins. The protein NpmA modifies the ribosomal RNA in a wide range of bacteria to prevent binding of aminoglycoside antibiotics. NpmA is a conserved methyltransferase that uses the cofactor S-adenosyl-L-methionine (SAM) to change the conformation of bacterial rRNA A1408 and induce methylation. This crucial step inhibits the binding of aminoglycosides to the bacterial rRNA. Four key lysine residues on the β2/3 linker region of NpmA are critical  to the recognition of rRNA as an appropriate substrate. The β5/6 linker of NpmA changes conformation upon binding to rRNA, which helps shift the position of E146. This change enables E146 to hydrogen bond to R207 in the β6/7 region, which keeps R207 in place. By forcing the location of R207, A1408 flips position to avoid steric hinderance. Once inverted, methylation of A1408 is induced by NpmA, which prevents binding of aminoglycosides to the bacterial rRNA, thus leading to resistance. Eliciting the role of different regions in NpmA could allow for development of substances capable of inhibiting its efficacy. Understanding the mechanism behind aminoglycoside resistance may lead to medications or techniques that could combat the bacterial adaption and slow the spread of resistance within populations.
to the recognition of rRNA as an appropriate substrate. The β5/6 linker of NpmA changes conformation upon binding to rRNA, which helps shift the position of E146. This change enables E146 to hydrogen bond to R207 in the β6/7 region, which keeps R207 in place. By forcing the location of R207, A1408 flips position to avoid steric hinderance. Once inverted, methylation of A1408 is induced by NpmA, which prevents binding of aminoglycosides to the bacterial rRNA, thus leading to resistance. Eliciting the role of different regions in NpmA could allow for development of substances capable of inhibiting its efficacy. Understanding the mechanism behind aminoglycoside resistance may lead to medications or techniques that could combat the bacterial adaption and slow the spread of resistance within populations.
More photos from CREST Conversations




