CREST Teaching and Research Symposium – May 4, 2013
Connecting Researchers, Educators and Students (CREST) is an NSF-CCLI project in which researchers, undergraduate educators and undergraduate students work together to explore a research topic and create educational materials for use in the undergraduate classroom. This program is a spin-off of the highly successful SMART Team program, in which high school students work with a researcher to create a physical model of a protein being studied in the research lab; and the PALM Project, in which ancillary educational materials (animations, illustrations, Jmol tutorials, paper bioinformatics and toober folding activities) were created to supplement accurate 3D models of proteins.
Undergraduate students interact with a research lab to understand the focus of research in the laboratory. Students then design a model of the protein being investigated in the lab; a physical model of the protein is then built using 3D printing technology. The students then work closely with an undergraduate educator to develop ancillary educational materials, so that their protein model can be incorporated in the undergraduate classroom. An important component of these materials is the incorporation of ‘how do we know what we know?’ – or the experiments that were done to elucidate the findings conveyed in the activities – so that students in the classroom see science not merely as a collection of facts collected in their textbook
Symposium Program (arranged to create a booklet if printed double-sided)Alverno College
TALEs: The Latest in Programmable DNAses
Students:
- Becka Anton
- Sarah Jackson
- Jasmine Reed
Faculty Advisors:
- Carl Ball, Ph.D.
- Heather Mernitz, Ph.D.
Research Mentor:
- Aron Geurts, Ph.D., Medical College of Wisconsin
PDB file:
- 3ugm.pdb
Presentation:
Abstract:
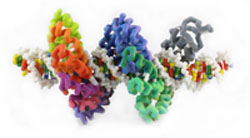
Transcription activator-like effectors (TALEs), which were originally isolated from a plant pathogen Xanthomonas, are high-affinity DNA binding proteins (Pennisi, 2012). These proteins have multiple 33 to 35-amino acid helix-loop-helix repeats, each with one variable amino acid pair in the loop that interacts with a specific nucleotide in the DNA helix. These repeat variable diresidues (RVDs) in the TALE protein make for a unique specificity, as there is a predictable pattern of correspondence between the RVD and the DNA base. The elucidation of this “code” allows the engineering of TALES that target specific genes. TALE fusion proteins offer the potential to target specific genes for DNA modification (Mak, 2012).
Concordia University Wisconsin
Modeling of the A6 T Cell Receptor Interacting with the Viral Tax Peptide
Students:
- Chris Bohl
- Joe Bok
Faculty Advisor:
- Ann McDonald, Ph.D.
Research Mentor:
- Amy Hudson, Ph.D., Medical College of Wisconsin
PDB file:
- 1ao7.pdb
Presentation:
Abstract:
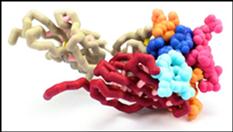
T-cell receptors (TCR) on cytotoxic T lymphocytes (CTL) detect the presence of intracellular infections by binding specifically to foreign peptides bound to class I major histocompatibility complex (MHC) on infected cells. TCRs are heterodimers consisting of alpha and beta chains found on the surface of T lymphocytes. The extracellular portions of these chains contain two domains, a constant and variable region. A constant transmembrane region found in both chains anchors the TCR to the T cell membrane. The variable domain contains complementarity determining regions (CDRs) composed of short stretches of amino acids that differ among TCRs. Each chain contains three CDRs that form a unique antigen binding site, which determines the specificity of the TCR to foreign peptides. The antigen binding site is created by random rearrangements of multiple, short gene fragments in the DNA. The modeled A6 TCR is specific for the Tax peptide of the human T-cell lymphotrophic virus type 1 (HTLV-1). The Tax peptide is composed of nine amino acids which interacts with the flat surface of the TCR. When the TCR binds the class I MHC-peptide complex, the CTL is activated. Activated CTLs eliminate intracellular infections by releasing substances which lyse the infected cell. Interaction between the TCR and class I MHC-peptide: complex is critical for eliminating the infection.
Concordia University Wisconsin – School of Pharmacy
Varenicline and the α4β2 Nicotinic Acetylcholine Receptor
Jmol Team:
- Joseph Holewa
- Veronica Juarez
- Thomas Puerzer
- Jacob Sutherland
Poster Team:
- Karena Creten
- Elizabeth Everett
- Sirr Grice
- Margaret Roesser
Faculty Advisors:
- Daniel Sem, Ph.D.
- Frank Dailey, M.S., Ph.D.
- Ernest Stremski, M.D., M.B.A.
PDB file:
- 4afg.pdb
Presentation:
Abstract:
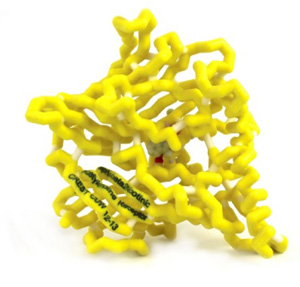
The addictive nature of nicotine is in part due to its high affinity for α4β2 neuronal nicotinic acetylcholine receptors (nAChR), which causes dopamine release in the brain producing the “reward” feeling of smoking. Varenicline, brand name Chantix©, is a partial agonist of the α4β2 nAChR (1). Analyzing the binding patterns of Varenicline to the receptor helps to understand the effectiveness of the drug in smoking cessation therapy. Varenicline and nicotine have similar molecular recognition involving water acting as a bridge within the binding pocket of the nAChR. Binding of a substrate to the 5 subunits of the nAChR causes a conformational change opening the pore of the receptor. Nicotine is a full agonist to the receptor while varenicline is a partial agonist. When the pore opens it allows ions into the nerve cell, which leads to depolarization, and release of dopamine. Varenicline relieves the craving and withdrawal symptoms because it releases some dopamine while simultaneously blocking the effects of nicotine by occupying the binding site; thus allowing patients to continue smoking during therapy, while simultaneously removing the “reward” that nicotine causes.
Saint Leo University
Alternate Splicing of the PKCδ Gene
Students:
- Robert Hartough
- Amanda Morris
- Jesse Robinson
- Umberto Napoletano
Faculty Advisor:
- Audrey Shor, Ph.D.
Research Mentor:
- Niketa Patel, Ph.D.
Presentation:
Abstract:
Protein Kinase C δ (PKCδ) is a member of a family of PKC isozymes that play significant roles in signal transduction pathways involving cell growth, differentiation and apoptosis. In humans, PKCδ may exist as one of two splice variants: PKCδ-I or PKCδ-VIII. The two variants have been implicated in cognition and Alzheimer’s disease, as well as dysfunctions related to obesity. Most notably, these variants perform opposing functions. PKCδ-I is responsible for apoptosis, whereas PKCδ-VIII promotes cellular survival. The drastic difference in function occurs through the alternative splicing of the PKCδ gene. Thirty-one additional amino acids are translated into the hinge region of PKCδ-VIII which prevents cleavage of the catalytic fragment that would normally promote apoptosis. Using PKCδ as an example, our goal in the immediate project was to develop an active learning tool that illustrates how alternative splicing can result in dramatic changes in protein structure and thus changes in protein function. Different models have been considered and beta testing has been conducted using an adapted version of one such model, and two other models have been prepared for testing and comparison. We believe an interactive RNA map will provide a memorable experience to help solidify the process of alternative splicing for novice learners.
Saint Leo University
Identification and Modeling of Conserved Secondary Structures of Influenza A Virus Hemagglutinin Subtypes H1, H2, H3, H5
Student:
- :Bradley Brooks
Faculty Advisors:
- Audrey Shor, Ph.D.
PDB files:
- 2wrc.pdb
- 2yp7.pdb
Presentation:
Abstract:
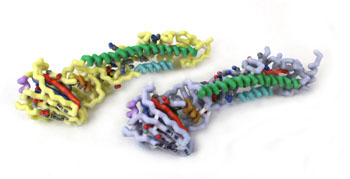
Influenza viruses cause epidemics and pandemics that result in millions of cases of severe illness and hundreds of thousands of deaths annually. Reassortments, interspecies transmission, and mutations lead to new strains with pathogenic potential. Influenza A viruses are responsible for the major human pandemics. Based on antigenic properties, influenza A viruses are classified into 17 hemagglutinin and 10 neuraminidase subtypes. Hemagglutinin is a surface glycoprotein that plays a critical role in host recognition, attachment, and entry. The globular head region of the protein is highly exposed and a major target of an immune response. This study investigates the structural similarities and difference between the HA1 peptide of H1, H2, H3, and H5 subtypes. Influenza A virus hemagglutinin protein sequences and crystallized structures available in the RCSB protein data bank, corresponding to H1, H2, H3, and H5 subtypes, were used to identify both highly variable and invariable regions of the glycoprotein. The HA1 peptide in the H1-H2, H5-H2, and H3-H2 comparison showed a 53.8, 68.3, and a 33.4% similarity, respectively. The structural analysis revealed an unaligned loop between H5 and H2 and a torsional twist between H3 and H2. Structured based approaches to drug discovery can utilize the 3D structures identified in this research for investigation novel therapies. Conserved regions of amino acids are resistant to mutations and can potentially be used as a target sites for neutralizing antibodies. These highly conserved regions have the potential to serve as the basis of a broad spectrum vaccine.
University of Wisconsin – Milwaukee
CheA-CheY Interactions in a Two Component System

Students:
- Trevor Croft
- Matthew Petri
- Jessica Schuld
Faculty Advisor:
- Steven Forst, Ph.D.
PDB file:
- 2lp4.pdb
Presentation:
Abstract:
Many bacteria respond to nutrient gradients by controlling the rotation of the flagella motor. Bacteria such as Escherichia coli sense environmental cues using a chemotaxis signal transduction system consisting of a kinase sensor, CheA and a response regulator, CheY. CheA undergoes autophosphorylation on a histidine residue and the phosphate group is in turn transferred to an aspartate residue on CheY. This study focuses on how CheA and CheY interact in order to provide correct orientation for the transfer of the phosphate group. CheA-CheY interactions have been elucidated by new NMR structures. The P1 and P2 functional domains located at the N-terminal region of CheA are involved in binding CheY. The P2 docking domain binds CheY with high affinity. Mutational analysis showed that Phe214 of the P2 domain is critical for high affinity binding. NMR analysis of the CheA-CheY complex revealed specific low affinity binding between CheY and P1. The interaction surface was predominantly with the first half of the α1 helix of P1. Phe8 and Glu15 of the α1 P1 play a role in specific binding of CheY to P1 CheA, as the rate of phosphorylation is 5-30 times slower when these residues have been mutated and rendered non-functional. These results refine our understanding of the structural features involved in CheA-CheY interactions and helps us to better appreciate the molecular mechanism of the phosphotransfer between the sensor kinase CheA and flagella motor-binding protein, CheY.