Campbell University (Buies Creek NC)
A Physical Model Overlay of Wild Type Influenza Virus A Neuraminidase and the His274Tyr Mutant Reveals the Basis of Drug Resistance and Catalysis
Team members: Abigaile Manzaro, Allison Blanchard, Travon Smith, Taylor Wilkins
Faculty advisor: Karen Guzman, Ph.D.
PDB IDs: Overlay1 of wild-type (2HU4) and H274Y mutant (3CL0) neuraminidases with the natural substrate, sialic acid (2BAT), and the drugs oseltamivir (3CL0) and zanamivir (2HTQ).
Abstract

The influenza virus is the causative agent of the flu, a contagious respiratory illness that causes morbidity, loss of productivity, and, in some instances, death. A strong arsenal of antiviral drugs is essential during a pandemic, but drug resistance threatens our preparedness. A key target for antivirals is neuraminidase, an enzyme that cleaves glycosidic bonds between sialic acid residues on cell surface glycoproteins so that the virus can infect or escape from host cells2. The challenges posed by the rapid evolution of drug resistance have led Dr. Celia Schiffer and her colleagues to develop more innovative ways, based on structural analyses, to formulate drugs that are impervious to resistance. Our focus is on the Oseltamivir resistance of His274Tyr neuraminidase mutant which has been shown to be due to a shift in the position of Glu276 into the hydrophobic pocket of the active site. We designed physical models based on the overlaid crystal structures of the wild type and the His274Tyr mutant to visualize the shift in Glu276 and its impact on binding of each of three potential ligands: Oseltamivir, Zanamivir, and Sialic Acid. In addition, the catalytic residues3 Tyr406 and Glu277 were displayed to visualize the lack of impact on catalytic cleavage of the natural substrate, but continued inhibition by Zanamivir. Our physical model of Neuraminidase shows active site residues to better visualize the basis of drug inhibition and resistance and illustrates a structural approach that may lead to a more effective method of drug design in the future.
Grand View University (Des Moines IA) – Dengue Team
Dengue NS32B Protease
Team members: Sarah Jordan, Aiden Martindale, Aaron Heuss, Dylan Miller, and Josh Tamse
Faculty advisor: Bonnie Hall, Ph.D.
PDB ID: 3u1j
Abstract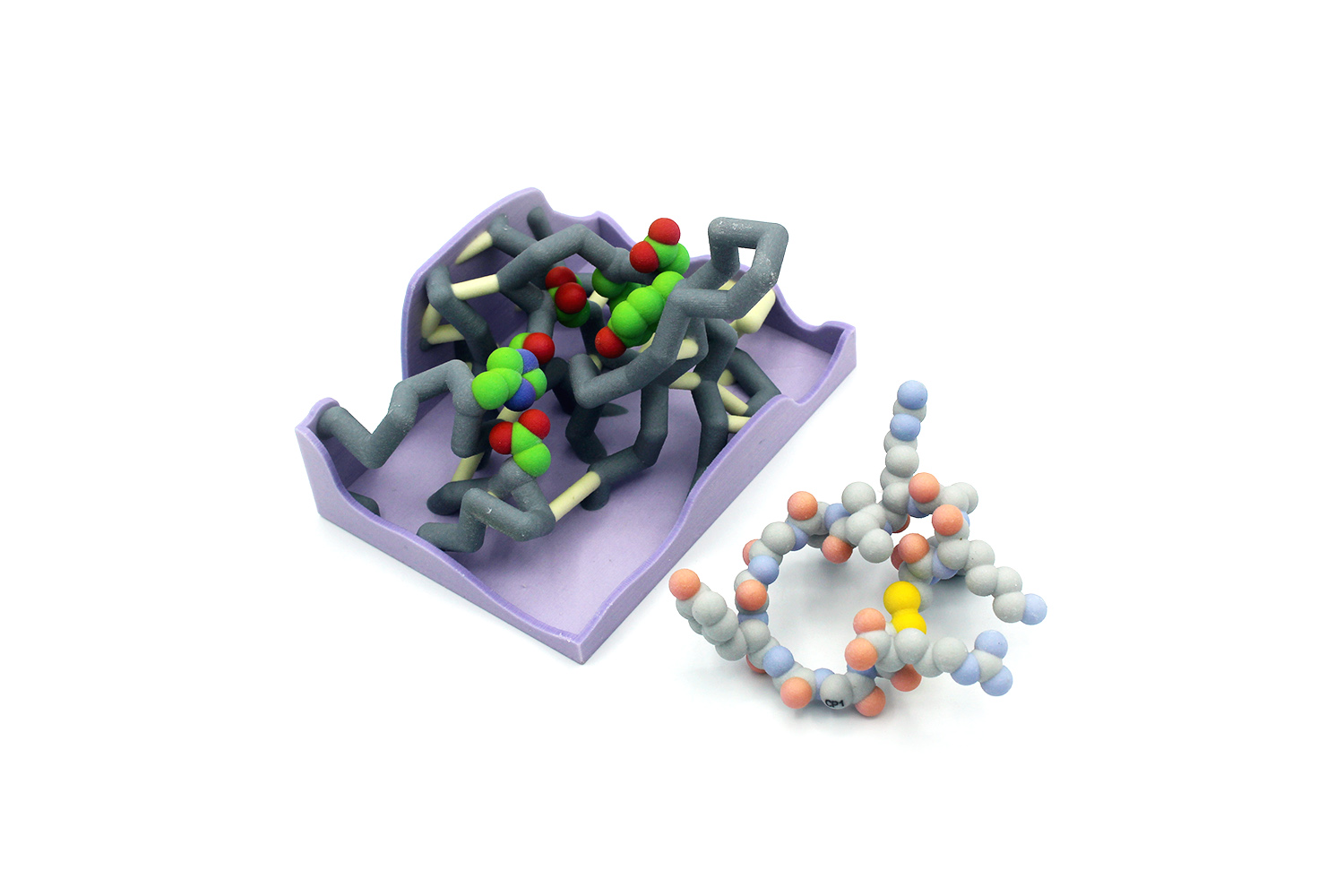
Annually, dengue fever affects an estimated 390 million people worldwide (more than the population of the entire United States). Symptoms include high fever, headache, vomiting, and muscle and joint pain so extreme it is also known as “break-bone fever”. Severe infection can lead to dengue hemorrhagic fever, dengue shock syndrome, and can be fatal. The disease is transmitted by mosquitos in tropical areas and is caused by the single-stranded RNA dengue virus (DENV). Currently the only treatment for patients infected with DENV is rest and hydration. The Schiffer lab has focused on the DENV NS2/NS3 serine protease as a potential drug target, because it is essential for DENV maturation. The serine protease inhibitor aprotinin (CP1) was the starting point for design of cyclic peptide (CP) inhibitors targeting DENV protease. A series of CP-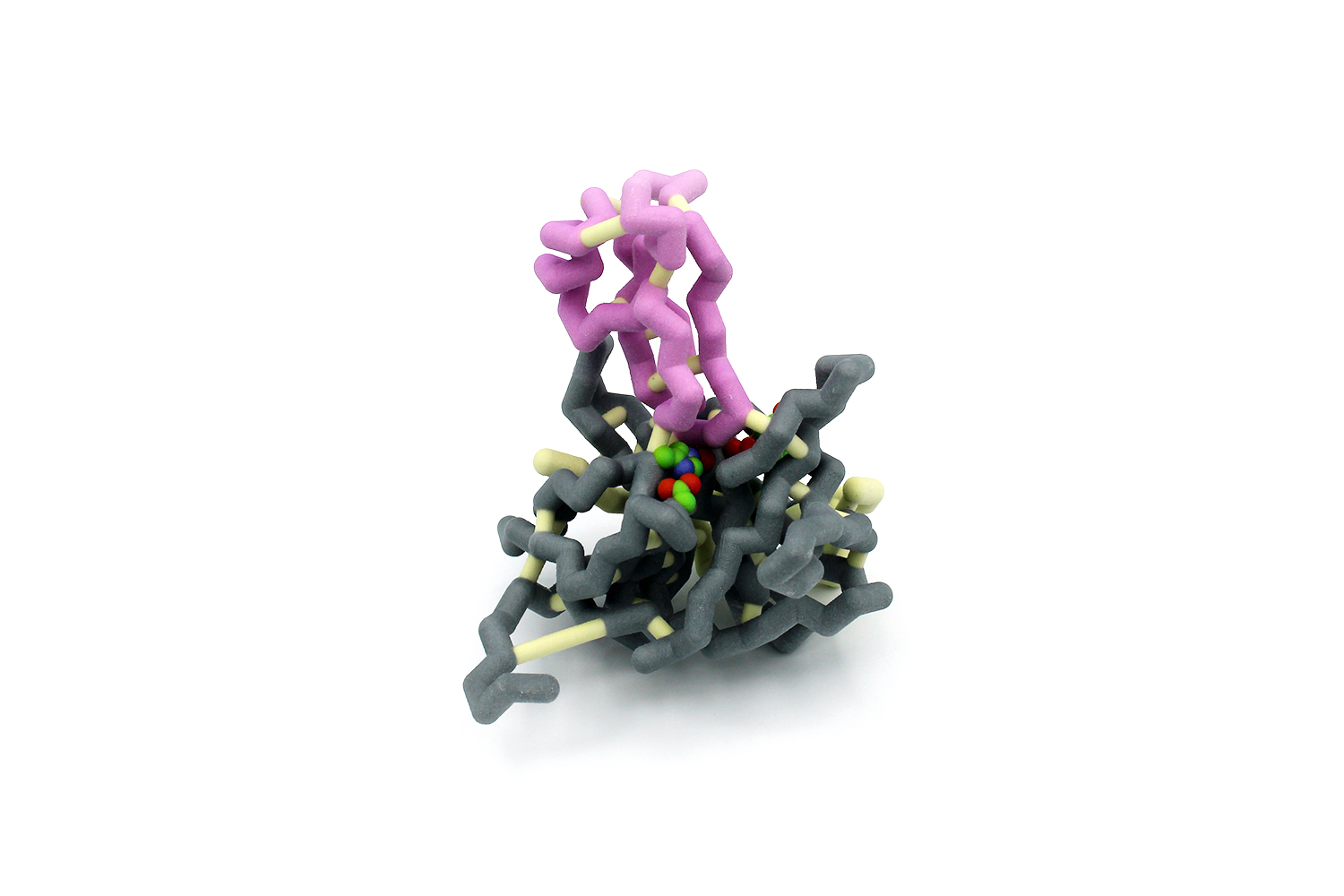 inhibitors were designed and tested, each with varying chemical properties. We designed a physical model of the DENV protease active site that displays the interactions with six different CP-inhibitors. The model demonstrates how CP inhibitor binding is impacted by drug size, flexibility, polarity, and ability to form intermolecular interactions (Lipinski’s Rule of Five). Our model also highlights how drug structure drives drug function and connects meaningfully to undergraduate studies of biology and chemistry at many levels. The DENV protease active site model, with the set of six CP inhibitors, is a useful classroom tool to show how basic biological and chemical principals are relevant to modern drug design.
inhibitors were designed and tested, each with varying chemical properties. We designed a physical model of the DENV protease active site that displays the interactions with six different CP-inhibitors. The model demonstrates how CP inhibitor binding is impacted by drug size, flexibility, polarity, and ability to form intermolecular interactions (Lipinski’s Rule of Five). Our model also highlights how drug structure drives drug function and connects meaningfully to undergraduate studies of biology and chemistry at many levels. The DENV protease active site model, with the set of six CP inhibitors, is a useful classroom tool to show how basic biological and chemical principals are relevant to modern drug design.
Grand View University (Des Moines IA) -Flu Crew
Structure of H1 1918 Hemagglutinin with Human Receptor
Team members: Lucas Burke, Karshana Kalyanaraman, Danielle Selover, Trang Khong, Stephanie Flores
Faculty advisor: Laura Salazar, Ph.D.
PDB ID: 2wrg
Abstract
 The Influenza A virus (IAV) causes a common and potentially fatal infection. The Center for Disease Control reports that 3-11% of the U.S. population is affected annually by IAV. The 2017-2018 burden of influenza led to over 950,000 patient hospitalizations and almost 80,000 deaths. The surface of IAV contains two proteins essential for infection: hemagglutinin (HA) and neuraminidase (NA). HA binds to a receptor on an epithelial host cell surface, and helps IAV penetrate the cell and release the viral RNA genome. As newly formed IAV particles leave the infected cell, NA assists in viral release and driving the infection of neighboring cells. Due to frequent, natural mutations in the IAV genome, annual IAV vaccines are engineered but are not always effective. The Schiffer Lab propagated IAV in vitro in the presence of an antibody to identify mutations of IAV that can escape immune detection. A critical asparagine located in the binding pocket of HA (N203HA) facilitates a hydrogen bond to sialic acid on LSTc (a human receptor analog). The N203VHA escape mutation disrupts the virus-ligand interaction; however, an allosteric mutation on NA (E329KNA) can compensate for the HA mutation. Our 3-D printed model of the HA active site visually depicts the binding of the N203 residue with the LSTc. This model further illustrates that viruses can use allosteric mutations to circumvent the immune system. Physically modeling the active site of HA allows students to investigate how substrates fit into active sites and visualize mutations within proteins.
The Influenza A virus (IAV) causes a common and potentially fatal infection. The Center for Disease Control reports that 3-11% of the U.S. population is affected annually by IAV. The 2017-2018 burden of influenza led to over 950,000 patient hospitalizations and almost 80,000 deaths. The surface of IAV contains two proteins essential for infection: hemagglutinin (HA) and neuraminidase (NA). HA binds to a receptor on an epithelial host cell surface, and helps IAV penetrate the cell and release the viral RNA genome. As newly formed IAV particles leave the infected cell, NA assists in viral release and driving the infection of neighboring cells. Due to frequent, natural mutations in the IAV genome, annual IAV vaccines are engineered but are not always effective. The Schiffer Lab propagated IAV in vitro in the presence of an antibody to identify mutations of IAV that can escape immune detection. A critical asparagine located in the binding pocket of HA (N203HA) facilitates a hydrogen bond to sialic acid on LSTc (a human receptor analog). The N203VHA escape mutation disrupts the virus-ligand interaction; however, an allosteric mutation on NA (E329KNA) can compensate for the HA mutation. Our 3-D printed model of the HA active site visually depicts the binding of the N203 residue with the LSTc. This model further illustrates that viruses can use allosteric mutations to circumvent the immune system. Physically modeling the active site of HA allows students to investigate how substrates fit into active sites and visualize mutations within proteins.
Hamline University (St. Paul MN)
HIV Protease
Team members: Sherina Dyrma, Cyndi Van Acker, Asha Nooh and Alessia Palacios
Faculty advisor: Betsy Martinez-Vaz, Ph.D.
PDB IDs: 6ops (active site box), 6dif (protein)
Abstract
 HIV infections affect over 38 million people worldwide and cause approximately one million deaths every year. Despite advances in antiretroviral therapies and disease prevention strategies, drug resistance remains a major challenge in HIV treatment. HIV protease plays a key role in viral replication by facilitating the peptide bond hydrolysis reactions necessary for the development of functional viral particles. Diverse inhibitors have been developed to target the active site and substrate envelope of HIV protease. While these drugs have been effective at inactivating the enzyme, mutations at the active site give rise to HIV resistance strains which are insensitive to protease inhibitors. The goal of this project was to create 3D printed models of HIV protease to illustrate the mechanisms of action of two inhibitors, Tipranavir and Darunavir, and the mutations involved in the development of resistance to these drugs. Database searches and sequence alignments were performed to identify conserved amino acids and structural features important in the catalytic mechanism of HIV proteases. Details of the protein structure and its interaction with Tipranavir and Darunavir were obtained by analyzing the Protein Databank Files: 6DIF, 6DGX, 6OPS, and 6DH6. To construct the physical models, the structure files were imported into Jmol and modified into
HIV infections affect over 38 million people worldwide and cause approximately one million deaths every year. Despite advances in antiretroviral therapies and disease prevention strategies, drug resistance remains a major challenge in HIV treatment. HIV protease plays a key role in viral replication by facilitating the peptide bond hydrolysis reactions necessary for the development of functional viral particles. Diverse inhibitors have been developed to target the active site and substrate envelope of HIV protease. While these drugs have been effective at inactivating the enzyme, mutations at the active site give rise to HIV resistance strains which are insensitive to protease inhibitors. The goal of this project was to create 3D printed models of HIV protease to illustrate the mechanisms of action of two inhibitors, Tipranavir and Darunavir, and the mutations involved in the development of resistance to these drugs. Database searches and sequence alignments were performed to identify conserved amino acids and structural features important in the catalytic mechanism of HIV proteases. Details of the protein structure and its interaction with Tipranavir and Darunavir were obtained by analyzing the Protein Databank Files: 6DIF, 6DGX, 6OPS, and 6DH6. To construct the physical models, the structure files were imported into Jmol and modified into  a format suitable for 3D printing using scripts created by undergraduate researchers. The 3D models feature the overall structure of HIV protease, and its inhibitor molecules, Tipranavir and Darunavir. An active site box was constructed to illustrate the biochemical interactions between the enzyme and the inhibitors. The physical models also highlight key amino acids such as Asp25, Ile50, Val82, and Val84, which are critical for catalytic activity and the development of drug resistance. Magnetic pieces were created to show the effects of mutations in inhibitor binding and enzyme activity. A Jmol tutorial was designed to complement the 3D models and assess students’ learning of the structure and function of HIV protease.
a format suitable for 3D printing using scripts created by undergraduate researchers. The 3D models feature the overall structure of HIV protease, and its inhibitor molecules, Tipranavir and Darunavir. An active site box was constructed to illustrate the biochemical interactions between the enzyme and the inhibitors. The physical models also highlight key amino acids such as Asp25, Ile50, Val82, and Val84, which are critical for catalytic activity and the development of drug resistance. Magnetic pieces were created to show the effects of mutations in inhibitor binding and enzyme activity. A Jmol tutorial was designed to complement the 3D models and assess students’ learning of the structure and function of HIV protease.
Lane College (Jackson TN)
HIV Protease
Team members: Keviyanah Gilmore, Alexis Craft, Tra’mya Lauderdale, Joy Jernigan, Ryan Billings
Faculty advisors: Melanie Van Stry, Ph.D. and Candace Jones, Ph.D.
PDB ID: 6dh8
Abstract
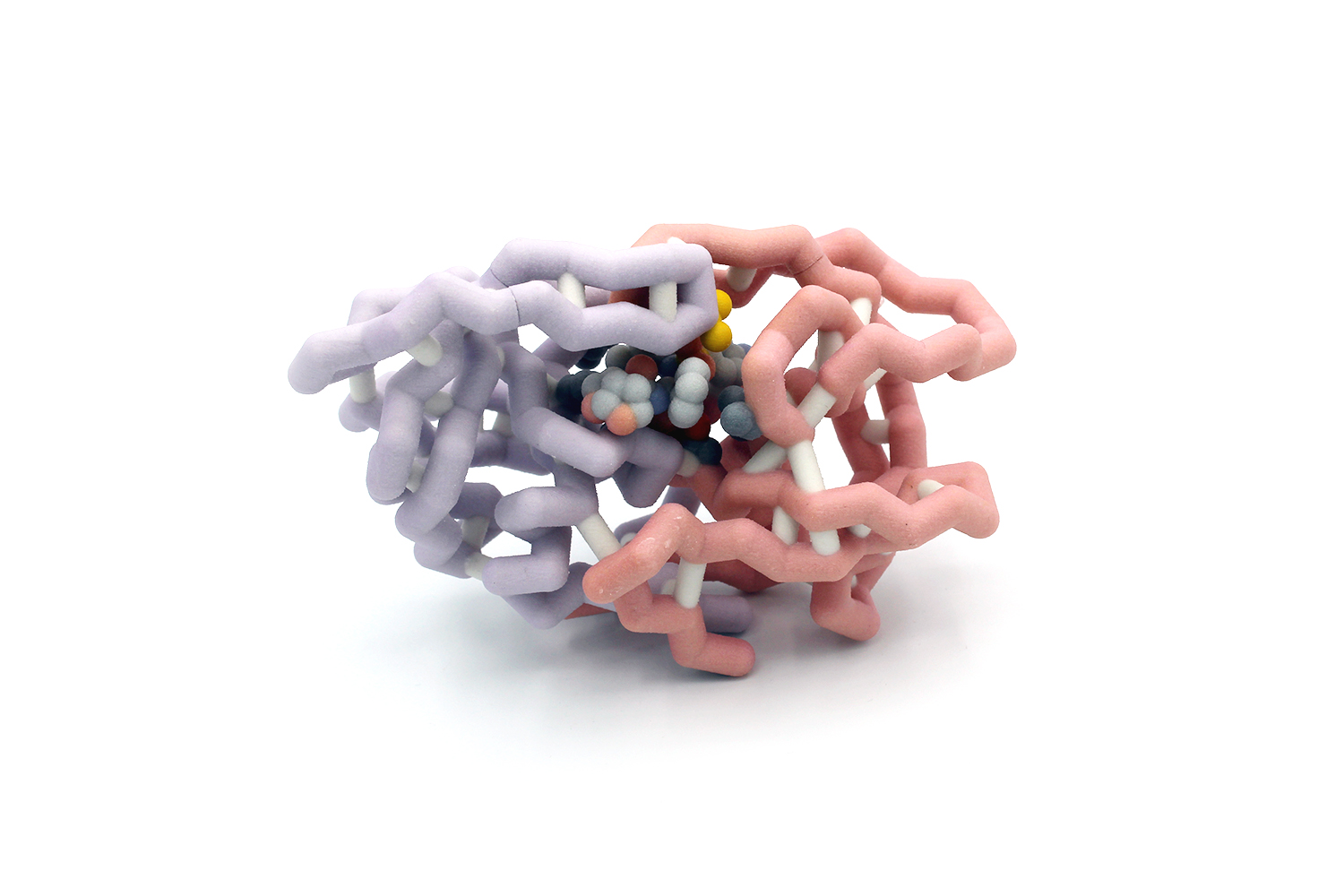 Human immunodeficiency virus (HIV) affects 37 million people worldwide. HIV causes Acquired Immunodeficiency Syndrome (AIDS) by impairing the immune functions in the human body and is transmitted through contact of body fluid. HIV attacks the body’s immune system, specifically CD4 T cells which help the immune system fight off infection. Antiretroviral therapy (ART) has prolonged the life expectancy of those under the treatment process. ART is a combination of antiretroviral drugs that keep the virus from replicating. The drug combination “cocktail” combines the power of multiple drugs to block multiple targets in HIV replication. HIV-1 protease is an essential enzyme for the life cycle of HIV. Therefore, HIV-1 protease is an important target used in antiretroviral therapy. However, under the selective pressure of protease inhibitors, mutations may occur at the active site, crippling inhibitor binding and leading to drug resistance. Umass6 is an analog of the HIV protease inhibitor, Duranovir, with a bulky P1’ moiety that allows enhanced overall van der Waals interactions at the active site. Here, we design a 3D model of the drug resistant HIV protease mutant I50V bound to UMass6 using Jmol based on the crystal structure described by Gordon J. Lockbaum and colleagues (1). HIV protease is a homodimer with the active site located in the interface between the subunits. Our model highlights the location of the I50V mutation within the active site and the resulting structural changes that cause the loss of inhibitor potency. Our 3D model also highlights the critical residues involved in inhibitor binding and the catalytic residue, D25, within the active site. Designing 3D model exposes students to protein structure, mutations, and tools for understanding drug resistance.
Human immunodeficiency virus (HIV) affects 37 million people worldwide. HIV causes Acquired Immunodeficiency Syndrome (AIDS) by impairing the immune functions in the human body and is transmitted through contact of body fluid. HIV attacks the body’s immune system, specifically CD4 T cells which help the immune system fight off infection. Antiretroviral therapy (ART) has prolonged the life expectancy of those under the treatment process. ART is a combination of antiretroviral drugs that keep the virus from replicating. The drug combination “cocktail” combines the power of multiple drugs to block multiple targets in HIV replication. HIV-1 protease is an essential enzyme for the life cycle of HIV. Therefore, HIV-1 protease is an important target used in antiretroviral therapy. However, under the selective pressure of protease inhibitors, mutations may occur at the active site, crippling inhibitor binding and leading to drug resistance. Umass6 is an analog of the HIV protease inhibitor, Duranovir, with a bulky P1’ moiety that allows enhanced overall van der Waals interactions at the active site. Here, we design a 3D model of the drug resistant HIV protease mutant I50V bound to UMass6 using Jmol based on the crystal structure described by Gordon J. Lockbaum and colleagues (1). HIV protease is a homodimer with the active site located in the interface between the subunits. Our model highlights the location of the I50V mutation within the active site and the resulting structural changes that cause the loss of inhibitor potency. Our 3D model also highlights the critical residues involved in inhibitor binding and the catalytic residue, D25, within the active site. Designing 3D model exposes students to protein structure, mutations, and tools for understanding drug resistance.
Milwaukee School of Engineering (Milwaukee WI)
Influenza Neuraminidase N1 Subtle Active Site Mutations for Viral Drug Resistance
Team members: Guinevere Sieradzki, Anna DeBruine, Connor Minx, Sabrina Mierswa, Amber Sabin, Jessica Koprek, Victoria Perez, Krishna Persaud, Josephine Selvik, Behrgen Smith, Joshua Zieman, Muskan Kanungo, Alexandra Berkowicz, Bella Cabrera-Brito, Benjamin Pung
Faculty advisor: Anne Alexander, Ph.D.
PDB IDs: 2hu4, 3cl0
Abstract
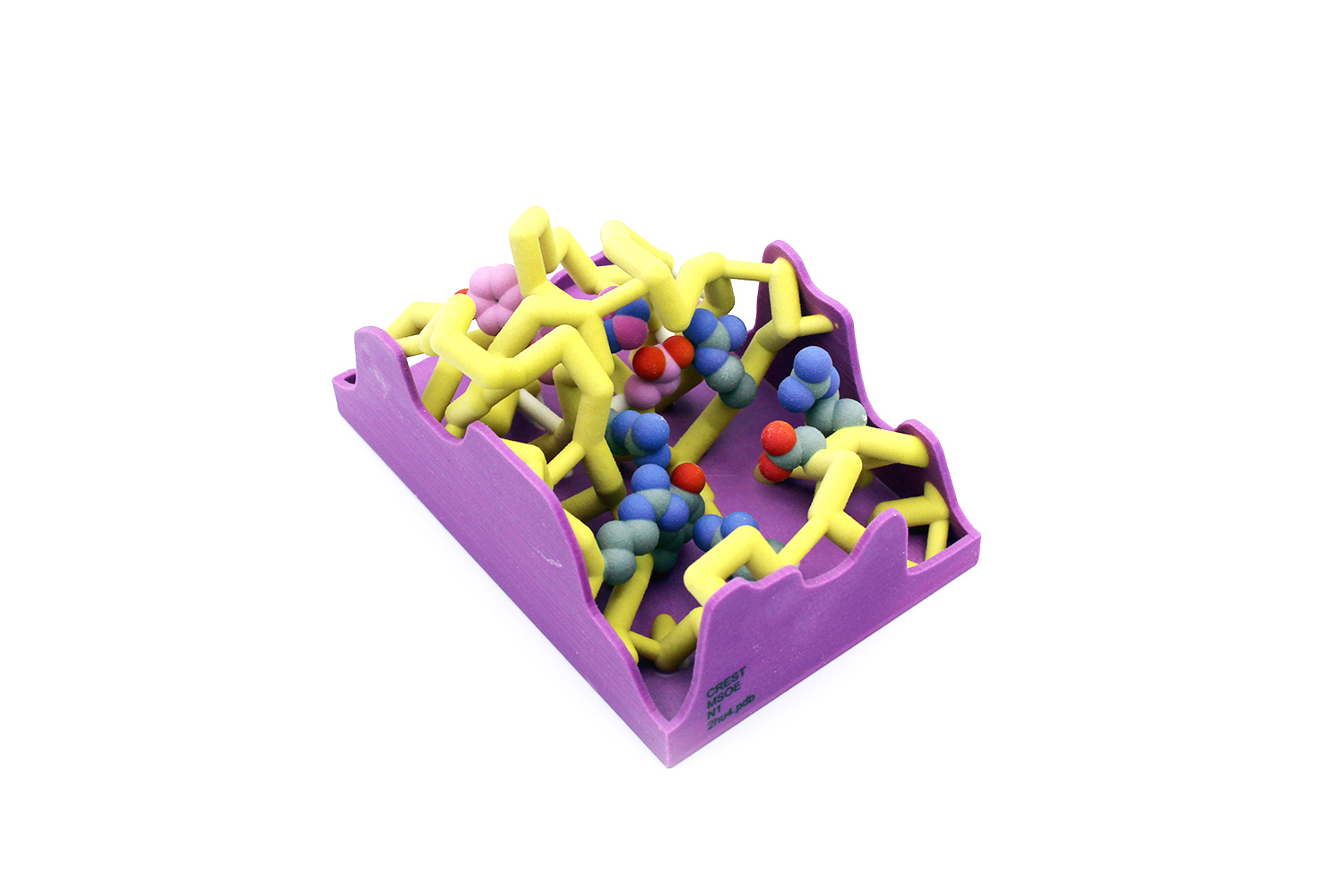 Influenza, also known as the “seasonal flu,” causes 24 million deaths annually. This number continues to rise because of the development of antiviral drug resistance within influenza types. The influenza virus is known for its rapid mutations, which may contribute to these resistance mechanisms. Neuraminidase is an important transmembrane protein on the surface of influenza and is an antiviral target. Viral neuraminidase (NA) cleaves sialic acid from the cell surface and progeny virions, causing the virus to release from infected cells. There are several subtypes of neuraminidase; this project focuses on neuraminidase 1 (N1) because of its prevalence in human influenza A infections. The catalytic residues on N1 are Arg118, Asp151, Arg152,
Influenza, also known as the “seasonal flu,” causes 24 million deaths annually. This number continues to rise because of the development of antiviral drug resistance within influenza types. The influenza virus is known for its rapid mutations, which may contribute to these resistance mechanisms. Neuraminidase is an important transmembrane protein on the surface of influenza and is an antiviral target. Viral neuraminidase (NA) cleaves sialic acid from the cell surface and progeny virions, causing the virus to release from infected cells. There are several subtypes of neuraminidase; this project focuses on neuraminidase 1 (N1) because of its prevalence in human influenza A infections. The catalytic residues on N1 are Arg118, Asp151, Arg152,  Arg224, Arg292, Arg371, and Tyr406. Oseltamivir (Tamiflu) is a common antiviral drug used to treat influenza A infections. It interacts with the active site of neuraminidase, functioning as a competitive inhibitor. N1 resistance to oseltamivir has arisen through a mutation at residue 274 in which the histidine (H) changes to a bulky tyrosine (Y). When this H274Y mutation occurs, alterations in the intermolecular contacts with the nearby glutamate at 276 push the amino acid further into the active site. The amino acids at positions 273 and 275 are also tyrosine residues. This creates a bulky network of residues in the active site, preventing the binding of oseltamivir. These conformational changes are subtle but comprise the central mechanism of viral drug resistance in N1. This mutation also causes the loss of nearby hydrogen bonds, further altering the stability and conformation of the active site.
Arg224, Arg292, Arg371, and Tyr406. Oseltamivir (Tamiflu) is a common antiviral drug used to treat influenza A infections. It interacts with the active site of neuraminidase, functioning as a competitive inhibitor. N1 resistance to oseltamivir has arisen through a mutation at residue 274 in which the histidine (H) changes to a bulky tyrosine (Y). When this H274Y mutation occurs, alterations in the intermolecular contacts with the nearby glutamate at 276 push the amino acid further into the active site. The amino acids at positions 273 and 275 are also tyrosine residues. This creates a bulky network of residues in the active site, preventing the binding of oseltamivir. These conformational changes are subtle but comprise the central mechanism of viral drug resistance in N1. This mutation also causes the loss of nearby hydrogen bonds, further altering the stability and conformation of the active site.
Nova Southeastern University (Fort Lauderdale FL)
Development of “Sharkavir”: A Hypothetical Inhibitor for HIV-1 Protease
Team members: Matthew Hunt, Feza Abbas, Lyla Abbas, Mina Ghali, Helana Ghali, Alesa Chabbra
Faculty advisors: Emily Schmitt, Ph.D. and Arthur Sikora, Ph.D.
PDB IDs: 1t3r (wild-type HIV-1 protease), 4dqf (Darunavir in complex with mutated HIV-1 protease, but only Darunavir was used), and 1N49 (Ritonavir)
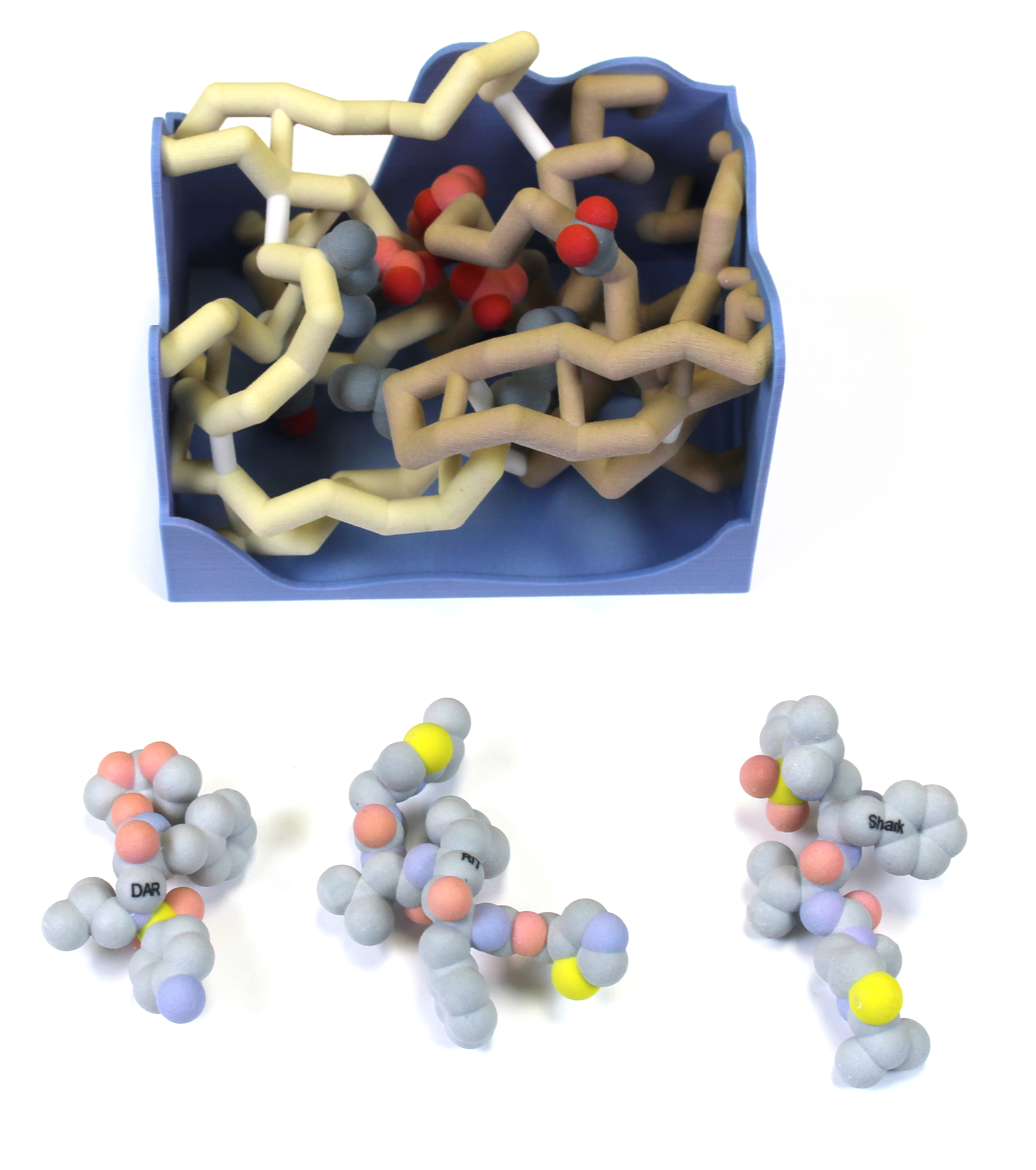 Abstract
Abstract
Human immunodeficiency virus (HIV) affects approximately 37 million people worldwide and results in over 1 million deaths annually. A class of drugs first developed in 1995 inhibits the enzyme HIV protease, thus preventing the maturation of an HIV precursor protein. There are now over 10 protease inhibitors available to treat HIV. Multiple mutations in the protein have made this treatment less effective (Mittal et al., 2012). The CREST (Connecting Researchers, Educators, and STudents) team at Nova Southeastern University modeled the interaction between HIV-1 protease and its inhibitors. Darunavir (DRV) is currently one of the strongest competitive inhibitors that exist as it binds effectively to the substrate envelope and has yielded a lower resistance for patients thus far. The most effective treatment is a combination of two of these inhibitors: Darunavir (DRV) and Ritonavir (RIT) (Mehta et al., 2019). In order to explain how these drugs work, the active site of the non-mutated wild-type HIV-1 protease was depicted as a binding box. The protease inhibitors Darunavir, Ritonavir, and our hypothetical drug, “Sharkavir”, were also 3-D printed to show how they fit into the protease active site (wild-type).
University of Wisconsin (La Crosse WI) – Team 1
HIV-1 Protease Bound to Darunavir
Team members: Bryton Foate, Ryan Maki, Lilly Riederer, and Rachel Senft
Faculty advisor: Basudeb Bhattacharyya, Ph.D.
PDB ID: 6opz
Abstract
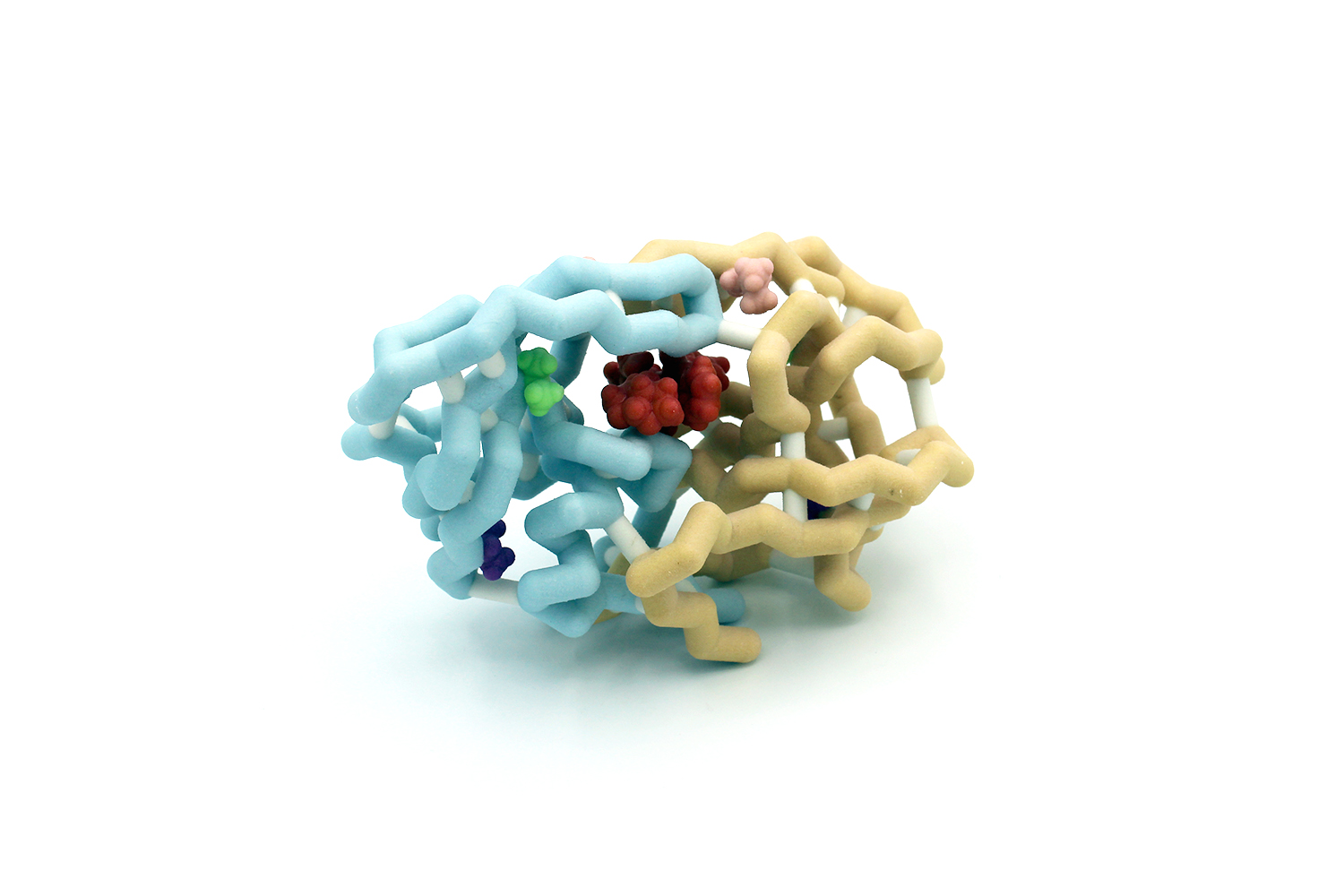 Pathogens such as bacteria and viruses are developing resistance faster than our ability to develop new drugs to fight them. This is creating a potentially grave problem for our society. HIV-1 is an example of a virus that has developed mechanisms to resist drugs. The virus requires a protease to cleave proteins in order for it to propagate. A highly effective drug that targets HIV-1 protease is darunavir, which works in nanomolar concentrations. The Schiffer group studied a series of amino acids commonly mutated in HIV-1 protease that confer drug resistance. Many of these residues are outside of the active site. We chose to model HIV-1 protease with mutations I54L (pink), A71V (purple), and L76V (light green) which are distal to the active site. These mutations together decrease darunavir’s effectiveness from 13 nM to 0.76 µM. The research provides implications in developing new drugs that could destroy HIV-1 as well as many other viruses that have a high mutation rate, such as the common cold or influenza.
Pathogens such as bacteria and viruses are developing resistance faster than our ability to develop new drugs to fight them. This is creating a potentially grave problem for our society. HIV-1 is an example of a virus that has developed mechanisms to resist drugs. The virus requires a protease to cleave proteins in order for it to propagate. A highly effective drug that targets HIV-1 protease is darunavir, which works in nanomolar concentrations. The Schiffer group studied a series of amino acids commonly mutated in HIV-1 protease that confer drug resistance. Many of these residues are outside of the active site. We chose to model HIV-1 protease with mutations I54L (pink), A71V (purple), and L76V (light green) which are distal to the active site. These mutations together decrease darunavir’s effectiveness from 13 nM to 0.76 µM. The research provides implications in developing new drugs that could destroy HIV-1 as well as many other viruses that have a high mutation rate, such as the common cold or influenza.
University of Wisconsin (La Crosse WI) – Team 2
Influenza Neuraminidase Chain A Bound to Zanamivir
Team members: Carter Caya, Gaoying Vue and Gina Wade
Faculty advisor: Basudeb Bhattacharyya, Ph.D.
PDB ID: 3b7e
Abstract
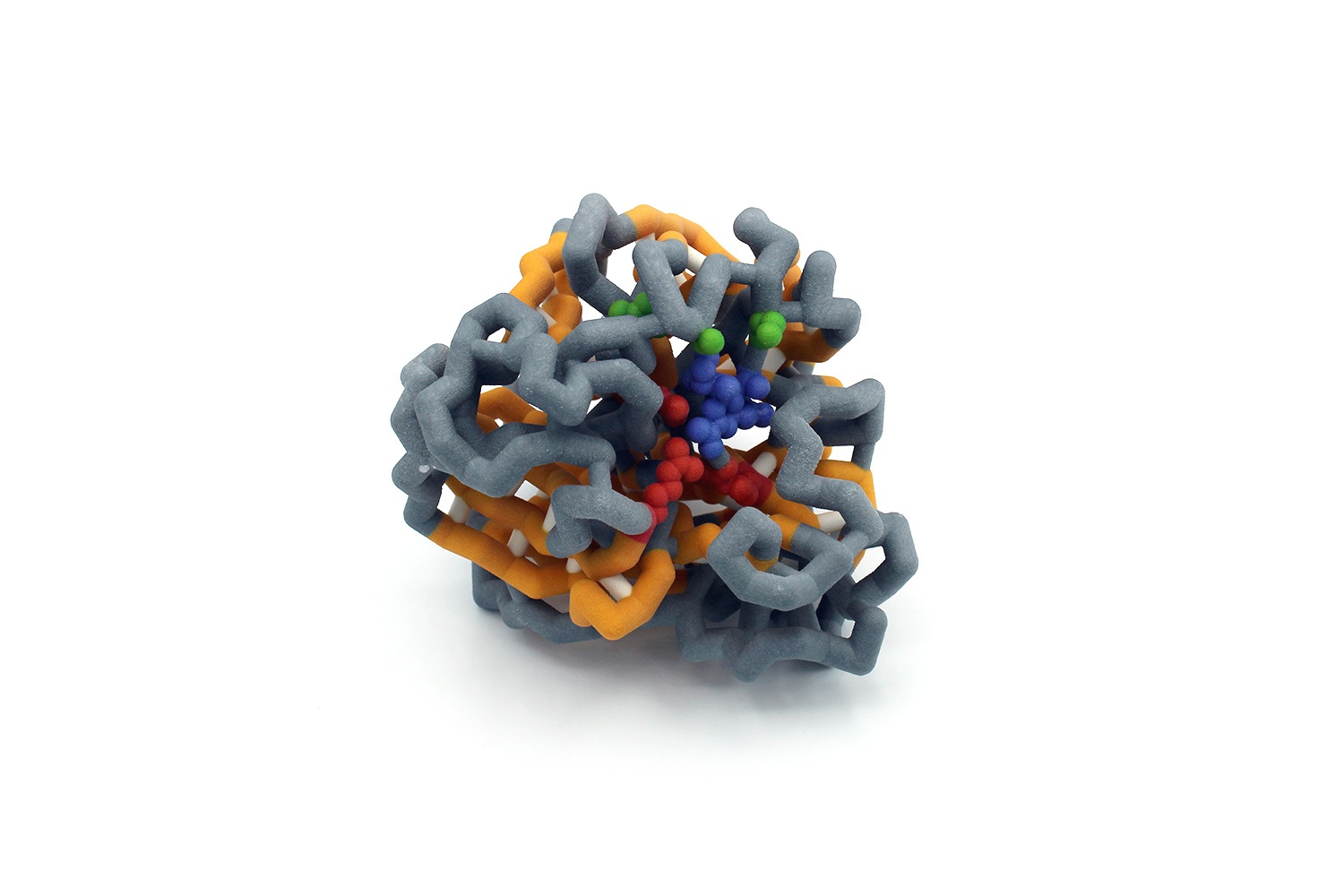 The 1918 influenza virus infected 25-30% worldwide killing at least 20 to 50 million people. While viral protein-targeting drugs have lessened the severity of present-day influenza infection, mutations have allowed the virus to remain elusive. The influenza protein neuraminidase is utilized to cut sialic acid from glycoproteins on host cell membrane, thus releasing the virus from the cell. The N1 neuraminidase subtype is prevalent in human infection and demonstrates a high mutations rate, conferring resistance to common viral drugs. Through hydrogen bonding, neuraminidase binds to its human substrate or inhibitor at amino acids E199, R118, R292, and R371 (red). Common N1 mutations occur at amino acids H274, I222, and S246 (green) which result in resistance to common neuraminidase inhibitors such as zanamivir. To understand how an inhibitor fits within N1, a three-dimensional model was built. Knowledge of the interactions between these mutated N1 amino acids and zanamivir will guide our understanding of influenza infection and help develop more effective treatments for drug-resistant strains.
The 1918 influenza virus infected 25-30% worldwide killing at least 20 to 50 million people. While viral protein-targeting drugs have lessened the severity of present-day influenza infection, mutations have allowed the virus to remain elusive. The influenza protein neuraminidase is utilized to cut sialic acid from glycoproteins on host cell membrane, thus releasing the virus from the cell. The N1 neuraminidase subtype is prevalent in human infection and demonstrates a high mutations rate, conferring resistance to common viral drugs. Through hydrogen bonding, neuraminidase binds to its human substrate or inhibitor at amino acids E199, R118, R292, and R371 (red). Common N1 mutations occur at amino acids H274, I222, and S246 (green) which result in resistance to common neuraminidase inhibitors such as zanamivir. To understand how an inhibitor fits within N1, a three-dimensional model was built. Knowledge of the interactions between these mutated N1 amino acids and zanamivir will guide our understanding of influenza infection and help develop more effective treatments for drug-resistant strains.
University of Wisconsin – Stevens Point
To Bind or Not to Bind: The Fickle Nature of HIV-1 Protease Inhibitors
Team members: Langenfeld, Noah J., Lang, Miranda L., Jiannacopoulos, Nina K., Augustus, Kaylie, Schultz, Tehya D., Devkota, Dipak, Korth, Cody R., Gulbronson, Chloe I., Corozolla, Rhiannon N., Goetsch, Quinn C., Chang, Keacha
Faculty advisor: Amanda Johnson, Ph.D.
PDB ID: 1t3r
Abstract
 Human Immunodeficiency Viruses (HIV) are detrimental RNA retroviruses that can lead to Acquired Immunodeficiency Syndrome (AIDS) in humans, causing over one million AIDS-related deaths each year. HIV-1 protease is a viral enzyme necessary for post-translational modification of the HIV proteins Gag and Gag-Pol. Inhibition of the protease’s substrate envelope has previously been a common target for developed drugs. However, the protease can easily mutate several residues including I84, V82, I50, and D25 within the envelope that confer resistance to common drug inhibitors. Efficient novel drug designs involve the modification of existing substrates to fit more tightly in the substrate envelope and maintain the ability to produce the desired result with and without mutated protease residues. Multiple analogs of darunavir (DRV), an FDA-approved inhibitor of HIV-1 protease, have been created with the slightly altered moieties of P1, P2, P1’ and P2’. In this project, we explore the interactions between the key protease residues and substrate components to better understand the binding strength of varying synthetic substrates. Increased knowledge of this system and its complexities could lead to smarter drug design choices in the future that remain usable in the face of frequent protease mutations. These new drugs could provide a more effective treatment for HIV positive individuals and help prevent the onset of AIDS, saving many lives and fighting back against constantly evolving proteases.
Human Immunodeficiency Viruses (HIV) are detrimental RNA retroviruses that can lead to Acquired Immunodeficiency Syndrome (AIDS) in humans, causing over one million AIDS-related deaths each year. HIV-1 protease is a viral enzyme necessary for post-translational modification of the HIV proteins Gag and Gag-Pol. Inhibition of the protease’s substrate envelope has previously been a common target for developed drugs. However, the protease can easily mutate several residues including I84, V82, I50, and D25 within the envelope that confer resistance to common drug inhibitors. Efficient novel drug designs involve the modification of existing substrates to fit more tightly in the substrate envelope and maintain the ability to produce the desired result with and without mutated protease residues. Multiple analogs of darunavir (DRV), an FDA-approved inhibitor of HIV-1 protease, have been created with the slightly altered moieties of P1, P2, P1’ and P2’. In this project, we explore the interactions between the key protease residues and substrate components to better understand the binding strength of varying synthetic substrates. Increased knowledge of this system and its complexities could lead to smarter drug design choices in the future that remain usable in the face of frequent protease mutations. These new drugs could provide a more effective treatment for HIV positive individuals and help prevent the onset of AIDS, saving many lives and fighting back against constantly evolving proteases.
Wabash College (Crawfordsville IN)
Exploring Drug Resistance in HIV-1 Protease using 3D Printed Models
Team members: Adan Villeda Jr., Austin Chivington, Maximilian Cobos, Thomas Gastineau, Cesar Mares, Javier Que
Faculty advisors: Walter R.P. Novak,Ph.D. and Lon A. Porter, Jr., Ph.D.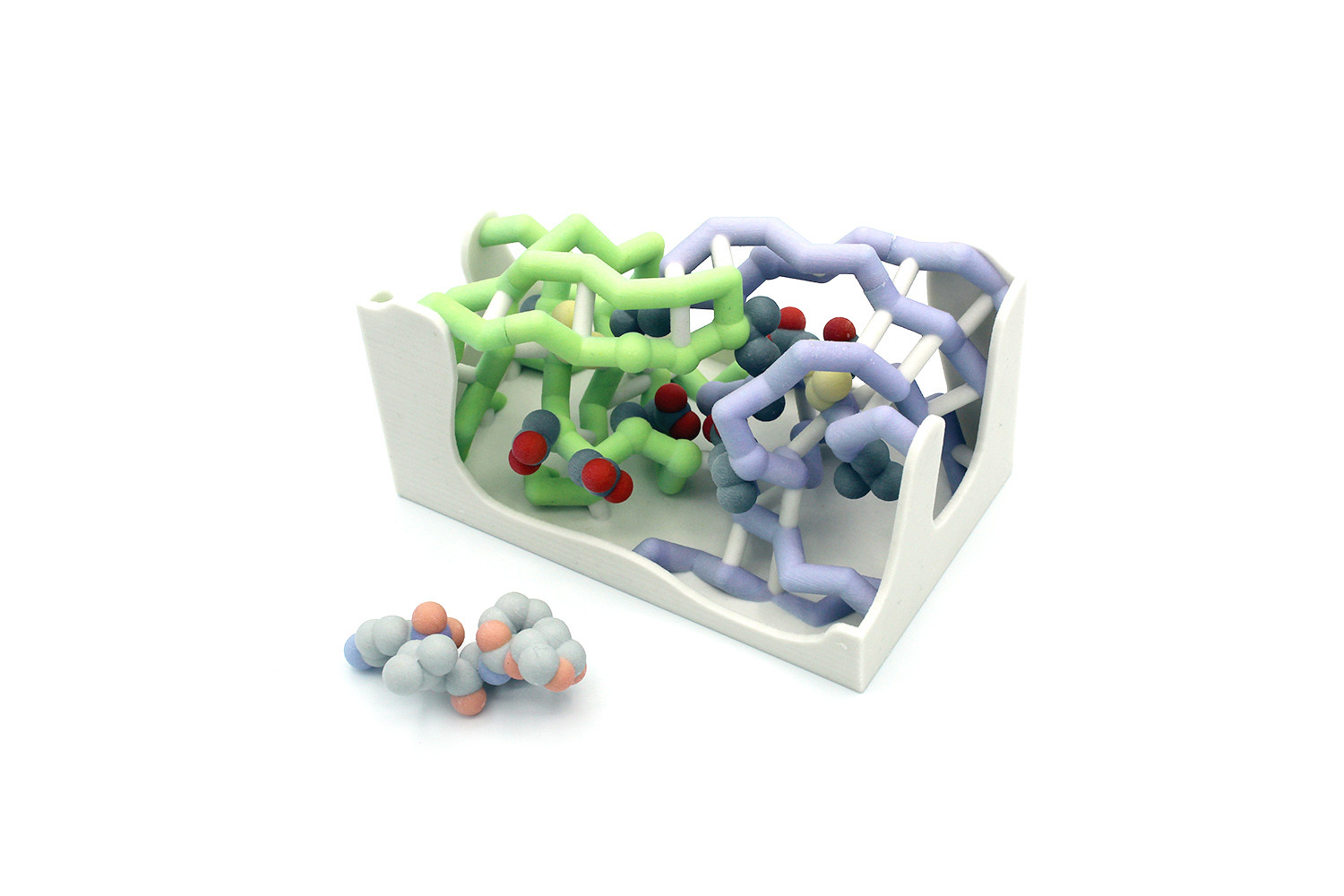
PDB ID: 4q1x with modifications
Abstract
HIV roughly affects 38 million people around the world. The enzyme HIV-1 protease is essential for replication and maturation of the virus. HIV-1 protease activates a variety of important proteins including reverse transcriptase and RNase H. Darunavir, an HIV-1 protease inhibitor, is the most potent antiretroviral drug to combat HIV to date. Despite the potency of Darunavir, HIV-1 protease can develop mutations both inside and outside of its active site to render Darunavir and other inhibitors ineffective. Here we develop 3D printed models to illustrate the structural basis of drug resistance. Specifically, we model the mutations V32I and L76V in HIV-1 protease. Hands-on models that illustrate resistance mutations near the active site of HIV-1 protease provide students with additional visual references to better understand drug resistance mechanisms and the design of novel inhibitors to effectively overcome these mutations.
Questions about the CREST Program? Contact crestprogram@gmail.com. We look forward to hearing from you!