Milwaukee School of Engineering (Milwaukee WI)
Crystal Structure of a Self-Splicing Group I Intron with Both Exons
Team members: R. Dohn, C. Ehrlich, M. Emch, G. Klas, J. Lang, Z. Paquin, I. Pavelich, A. Polovchak, L. Readnour, H. Steiner, P. Tebon, J. Verburgt and J. Wood
Faculty advisor:
PDB ID: 1u6b
Abstract
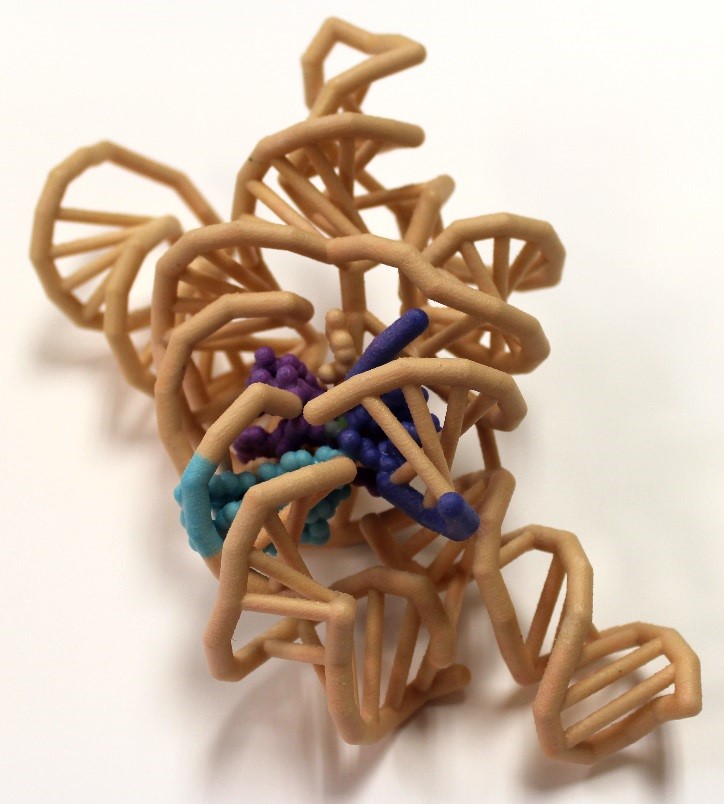
The discovery of catalytic RNA in Tetrahymena proved that not all enzymes are proteins. Messenger RNA (mRNA) splices out with the aid of proteins in a spliceosome complex. However, group I introns are self-splicing and are able to catalyze their removal from a primary RNA transcript without any additional accessory factors. The tertiary structure of the ribozyme places the highly reactive nucleotides into an active site containing metal ion cofactors. The crystal structure of Azoarcus sp. BH72 was visualized to understand the splicing mechanisms of the ribozyme. This crystal structure is the first to feature the junction between the intronic and exonic regions. It also features a set of two base triples which are stacked together at the active site, properly orienting the regions to be spliced. The intron’s O3′ displacement from the scissile phosphate by the 5′ -exon’s O3′ requires the unification of the 5′ and 3′ exons, and is likely catalyzed by the two metallic ion cofactors of the RNA that interact to help neutralize the charges in the dense phosphate region. The ribozyme requires at least two magnesium ions within its active site, activating the exon nucleophile, stabilizing the intron leaving group, and coordinating one of the metal ions to the ΩG O2′.
Mount Mary University (Milwaukee WI)
Phenylalanine Hydroxylase (PAH)
Team members: Alexis Cooper, Sarah Mahn and Andrea Roell
Faculty advisors: Maureen Leonard, Ph.D. and Colleen Conway, Ph.D.
PDB ID: 1j8b
Abstract
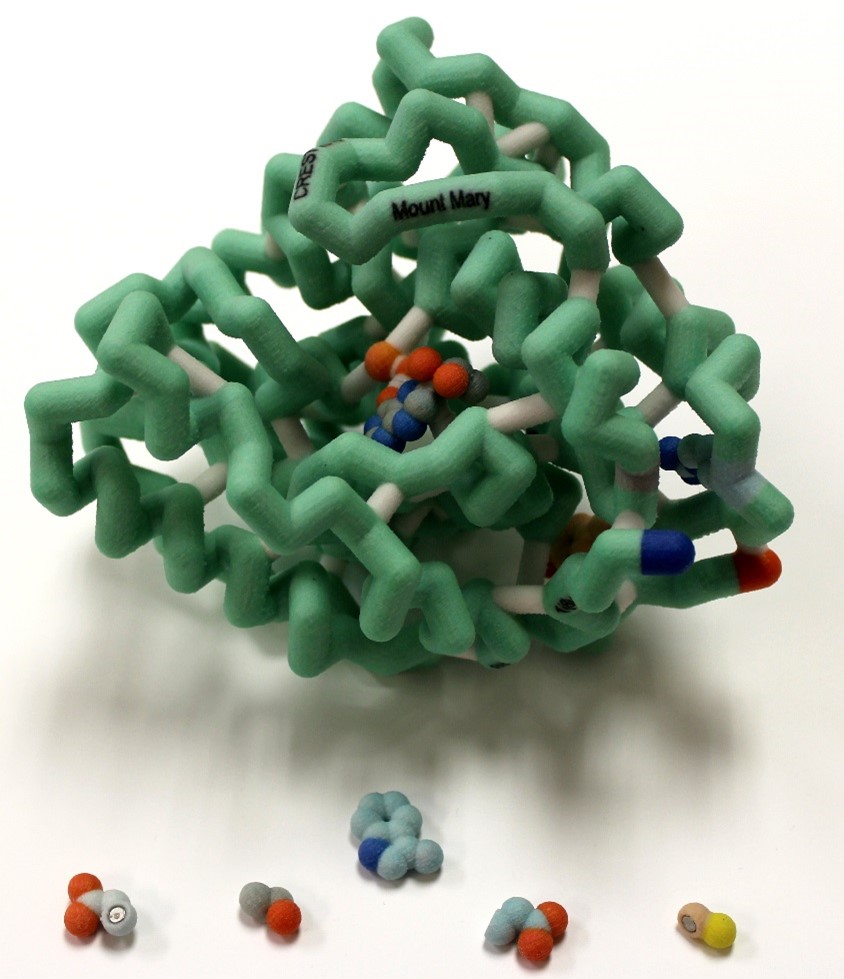 Phenylketonuria (PKU) is an autosomal recessive disorder of the phenylalanine hydroxylase gene (PAH). PKU results from mutations to the gene for this enzyme and is a common metabolic disorder affecting approximately 1 in 10,000 births. There are over 600 known mutations. The cofactor tetrahydrobiopterin (BH4) is needed for catalysis to occur. For individuals with PKU, the traditional treatment is a very restrictive low phenylalanine/low protein diet, but drug treatment with a BH4 analog, sapropterin, is a prospective treatment for patients to increase residual activity of PAH. We selected four point mutations in the catalytic domain that respond to BH4 treatment. A 3D model of the catalytic domain displaying the sites of these mutations (and magnet-docked wild type and mutant residues) and a gene map were constructed to visualize the location of these selected residues and hypothesize their importance for stabilizing secondary structure, as well as analyze their location within the gene. Analyzing the mutations involved can help predict which patients would likely respond to BH4 treatment, allowing for a more flexible diet.
Phenylketonuria (PKU) is an autosomal recessive disorder of the phenylalanine hydroxylase gene (PAH). PKU results from mutations to the gene for this enzyme and is a common metabolic disorder affecting approximately 1 in 10,000 births. There are over 600 known mutations. The cofactor tetrahydrobiopterin (BH4) is needed for catalysis to occur. For individuals with PKU, the traditional treatment is a very restrictive low phenylalanine/low protein diet, but drug treatment with a BH4 analog, sapropterin, is a prospective treatment for patients to increase residual activity of PAH. We selected four point mutations in the catalytic domain that respond to BH4 treatment. A 3D model of the catalytic domain displaying the sites of these mutations (and magnet-docked wild type and mutant residues) and a gene map were constructed to visualize the location of these selected residues and hypothesize their importance for stabilizing secondary structure, as well as analyze their location within the gene. Analyzing the mutations involved can help predict which patients would likely respond to BH4 treatment, allowing for a more flexible diet.
Saint Leo University (Saint Leo FL)
Tracing Base Mutations in PRPc Through Molecular Modeling to Aid in Determining Mutational Effects on Alzheimer’s Disease
Team member: Elizabeth Panek
Faculty advisor: Audrey Shor, Ph.D.
PDB ID: 1qm2
Abstract
 The conformational change in prion hPrPc to hPrPsc has been known to have detrimental effects on the human body, specifically neurological disorders such as Alzheimer’s. In addition, hPrPc has been known to bind to copper. These interactions have been speculated to have an impact on hPrPc, hPrPsc, and the pathways that they are theoretically involved in. Specifically, hPrPc has been thought to interfere with the FYN pathway. The speculated concept that is to be analyzed specifically is the relationship to copper and hPrPc as that to stabilize the raft for shuttling of amyloid beta in the pathway. Without this, it is theorized that both amyloid beta and hPrPc aggregate, which causes the effects seen in neurological disorders. This is theorized by the fact that dimers of amyloid beta can bind to hPrPc and activates PKY2. PKY2 then can go on to phosphorylate FYN. This causes the hyperphosphorylation of tau into ptau. Amyloid beta plaques and neurofibrillary tangles caused by the presence of ptau are common symptoms of Alzheimer’s disease, one of the diseases that hPcPsc is being considered as the cause of. Thus this project hopes to specifically look at this theorized relationship between Alzheimer’s disease and the isoforms of hPrPc and hPrPsc. Modern research is currently being done on how to block the hPrPc amyloid beta binding to treat Alzheimer’s patients. One interesting observation that is of high importance is that mutations G127V and M129V will prevent the development of neurological disorders caused by PrPc. Thus, this study investigates the impact of mutations G127V and M129V on copper binding stability of hPrP subunit 1QM2.
The conformational change in prion hPrPc to hPrPsc has been known to have detrimental effects on the human body, specifically neurological disorders such as Alzheimer’s. In addition, hPrPc has been known to bind to copper. These interactions have been speculated to have an impact on hPrPc, hPrPsc, and the pathways that they are theoretically involved in. Specifically, hPrPc has been thought to interfere with the FYN pathway. The speculated concept that is to be analyzed specifically is the relationship to copper and hPrPc as that to stabilize the raft for shuttling of amyloid beta in the pathway. Without this, it is theorized that both amyloid beta and hPrPc aggregate, which causes the effects seen in neurological disorders. This is theorized by the fact that dimers of amyloid beta can bind to hPrPc and activates PKY2. PKY2 then can go on to phosphorylate FYN. This causes the hyperphosphorylation of tau into ptau. Amyloid beta plaques and neurofibrillary tangles caused by the presence of ptau are common symptoms of Alzheimer’s disease, one of the diseases that hPcPsc is being considered as the cause of. Thus this project hopes to specifically look at this theorized relationship between Alzheimer’s disease and the isoforms of hPrPc and hPrPsc. Modern research is currently being done on how to block the hPrPc amyloid beta binding to treat Alzheimer’s patients. One interesting observation that is of high importance is that mutations G127V and M129V will prevent the development of neurological disorders caused by PrPc. Thus, this study investigates the impact of mutations G127V and M129V on copper binding stability of hPrP subunit 1QM2.