
2020-2021 CREST Teams
Campbell University (Buies Creek NC)
A Physical Model to Visualize the Unique SARS-CoV-2 ORF8 Dimerization Interfaces Potentially Critical to Pathogenesis
Team members: Bailey Hart, Ryan Peterson, Allison Robinson, Leeann Stearns, Ryan Vazquetelles, Elizabeth Wiles
Faculty advisor: Karen Guzman, Ph.D.
Introductory Video
Project Overview
The Campbell University Team has been exploring the ORF8 protein from SARS-CoV-2. They have been intrigued by the suggestion that this protein, which is a dimer, might form higher order structures that may derail the host immune response (Flower et al., 2020). They have contacted the researchers and are working with a postdoc from the lab (Cosmo Buffalo, Ph.D.) to create a higher order structure.
PDB IDs: unpublished files
Abstract
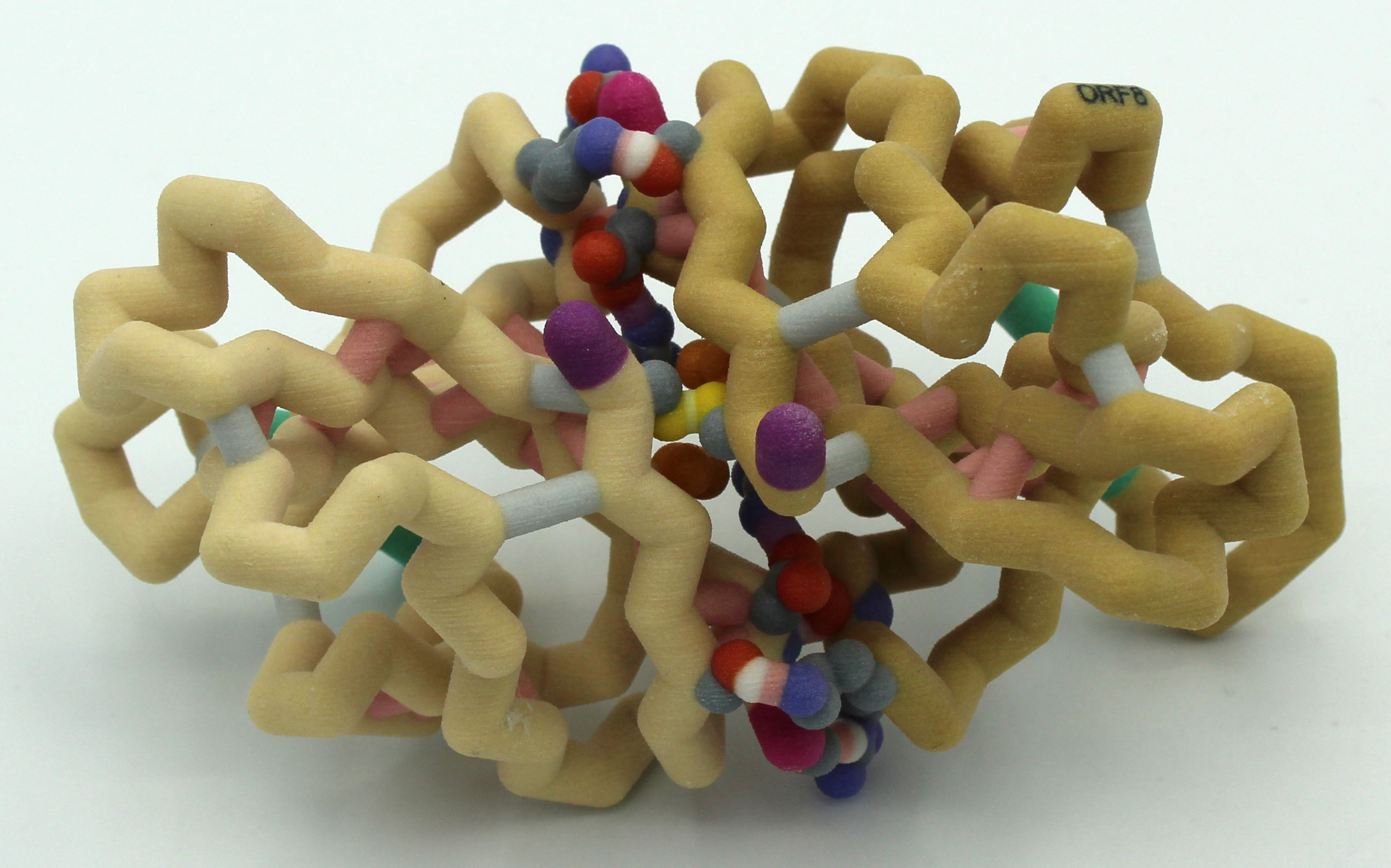
As the world continues to fight the coronavirus pandemic caused by SARS-CoV-2, we are gaining valuable insights by comparing this virus to other human coronaviruses such as SARS-CoV and MERS-CoV that have also caused outbreaks. Coronaviruses infect many animal species, but disease severity varies between strains. Viral accessory proteins are generally implicated in increased infectivity, pathogenicity and virulence. The accessory protein ORF8, although not essential for viral replication, appears to play a critical role in disease severity by inhibiting multiple pathways of the immune response. Furthermore, the ORF8 gene is found within a highly variable portion of the genome, increasing the possibility of dangerous mutant forms arising. Structure-based design of therapeutics holds great promise for effective targeting of such rapidly evolving threats, so the design 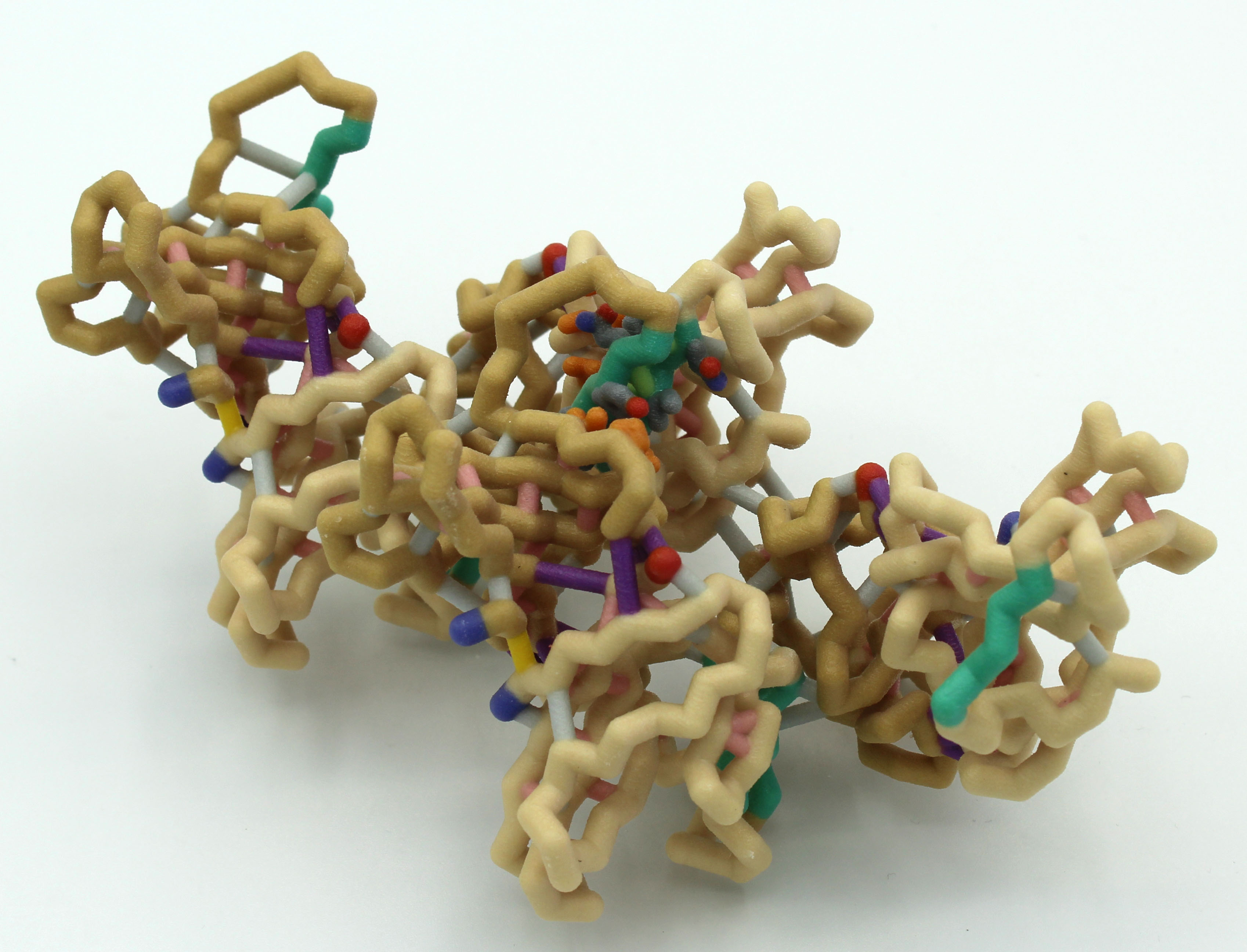 of physical models may improve our understanding of structural features responsible for pathogenicity and focus our efforts. ORF8 exists as a homodimer, i.e. two identical subunits, held together by amino acid residues in the dimerization domains that interact through hydrophobic interactions, hydrogen bonding, a salt bridge and a disulfide bond. Another dimerization interface, held together by an intermolecular beta sheet and stabilized by an extensive hydrophobic surface, exists between separate homodimers and results in oligomerization, i.e. formation of a complex composed of many homodimers. These two dimerization domains are unique to SARS-CoV-2 and have been proposed to be involved in the protein’s ability to evade and suppress the host immune response. Unique structural features such as these may be prime targets for development of therapeutics.
of physical models may improve our understanding of structural features responsible for pathogenicity and focus our efforts. ORF8 exists as a homodimer, i.e. two identical subunits, held together by amino acid residues in the dimerization domains that interact through hydrophobic interactions, hydrogen bonding, a salt bridge and a disulfide bond. Another dimerization interface, held together by an intermolecular beta sheet and stabilized by an extensive hydrophobic surface, exists between separate homodimers and results in oligomerization, i.e. formation of a complex composed of many homodimers. These two dimerization domains are unique to SARS-CoV-2 and have been proposed to be involved in the protein’s ability to evade and suppress the host immune response. Unique structural features such as these may be prime targets for development of therapeutics.
Grand View University (Des Moines IA) – Drug Design Team
SARS-CoV-2 Main Protease (nsp5)
Team members: Nick Buttolph, Danny Cavlovic, Dalton Dencklau, Michel Evertsen, Isabelle Juhler, Kallista Larson, Alexa Pinos, Jesse Rothfus
Faculty advisor: Bonnie Hall, Ph.D.
Project Overview
The Grand View University Drug Design Team is exploring drugs that bind to the main protease (nsp5, 3C-like protease) of SARS-CoV-2. They are interested in constructing a model of the whole protein with an existing drug bound, as well as an active site box, to which they can insert an existing drug, as well as drugs the team will design using computational approaches.
PDB ID: 6lu7
Abstract
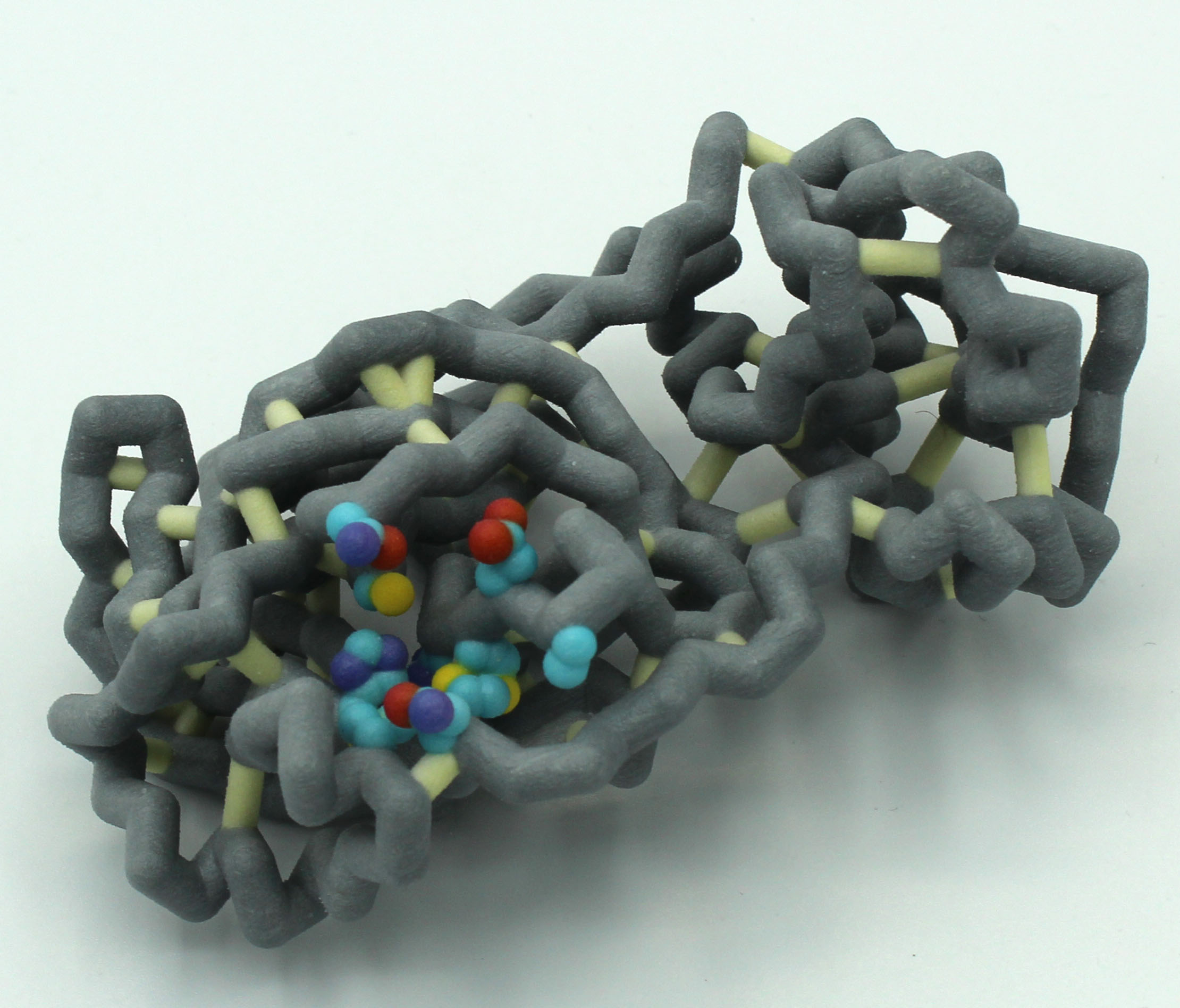 A new coronavirus causing an acute respiratory syndrome (SARS-CoV-2) emerged in late 2019 and has been responsible for the outbreak of coronavirus disease 2019 (COVID-19). Symptoms of COVID-19 range from mild to severe, including respiratory symptoms, systemic inflammatory responses, and even death. Currently, remdesivir is the only FDA-approved therapeutic agent to treat COVID-19, although it shows limited efficacy. We were interested in learning about the drug design process for a novel virus. We focused on drug research targeting the SARS-CoV-2 main protease, known as nsp5 or Mpro. The nsp5 protease is an enzyme critical for viral replication, with ebselen, cinanserin, and N3 being lead compounds identified as possible effective drugs (Jin, 2020). Starting with these chemical drug structures, we used an in silico drug design process to explore how to modify and refine these drugs. Our goal was to make the compounds more drug-like, according to Lipinski’s and Veber’s rules. Using the crystal structure file 6YB7 from the Protein Data Bank
A new coronavirus causing an acute respiratory syndrome (SARS-CoV-2) emerged in late 2019 and has been responsible for the outbreak of coronavirus disease 2019 (COVID-19). Symptoms of COVID-19 range from mild to severe, including respiratory symptoms, systemic inflammatory responses, and even death. Currently, remdesivir is the only FDA-approved therapeutic agent to treat COVID-19, although it shows limited efficacy. We were interested in learning about the drug design process for a novel virus. We focused on drug research targeting the SARS-CoV-2 main protease, known as nsp5 or Mpro. The nsp5 protease is an enzyme critical for viral replication, with ebselen, cinanserin, and N3 being lead compounds identified as possible effective drugs (Jin, 2020). Starting with these chemical drug structures, we used an in silico drug design process to explore how to modify and refine these drugs. Our goal was to make the compounds more drug-like, according to Lipinski’s and Veber’s rules. Using the crystal structure file 6YB7 from the Protein Data Bank  (PDB), we focused on the protease active site, defined by amino acids His41, Met49, Asn142, Cys145, His164, Met165, Glu166, Pro168, and Gln189. Cys145 is especially important, as it is the target for irreversible covalent inhibitors. Based on this information, we used molecular docking software to first re-dock these drugs into the nsp5 protease. We then made changes to the structures of the lead compounds, docked those new compounds, and used the energetic information to continue the refinement process. Once we completed our own drug design explorations, we created a three-dimensional printed model of the full nsp5 protease, with the key amino acids highlighted. We also created a three-dimensional model zoomed in on the active site, with the different drugs fitting into that active site model. Here, we present what we learned about the drug design process for a novel virus such as SARS-CoV-2. Our docking protocols and models are useful for teaching the fundamentals of drug design and about modern drug discovery and design processes.
(PDB), we focused on the protease active site, defined by amino acids His41, Met49, Asn142, Cys145, His164, Met165, Glu166, Pro168, and Gln189. Cys145 is especially important, as it is the target for irreversible covalent inhibitors. Based on this information, we used molecular docking software to first re-dock these drugs into the nsp5 protease. We then made changes to the structures of the lead compounds, docked those new compounds, and used the energetic information to continue the refinement process. Once we completed our own drug design explorations, we created a three-dimensional printed model of the full nsp5 protease, with the key amino acids highlighted. We also created a three-dimensional model zoomed in on the active site, with the different drugs fitting into that active site model. Here, we present what we learned about the drug design process for a novel virus such as SARS-CoV-2. Our docking protocols and models are useful for teaching the fundamentals of drug design and about modern drug discovery and design processes.
Grand View University (Des Moines IA) – RdRp Team
Structure of RdRp Complex and its Inhibition by Remdesivir
Team members: Sierra Dale, Asal Eid, Karshana Kalyanaraman, Binh Nguyen
Faculty advisor: Laura Salazar, Ph.D.
Introductory Video
Project Overview
The Grand View University RdRp Team has been exploring the work of Yin et al., looking at how Remdesivir inhibits the RNA-dependent RNA polymerase (RdRp; nsp12) of SARS-CoV-2. They are interested in building the RdRp showing interactions with accessory proteins nsp7 and nsp8. They also want to design a model of the double-stranded RNA, showing how Remdesivir, which is similar to adenine, blocks replication of RNA.
PDB ID: 7bv2
Abstract
 The SARS-CoV-2 virus, which causes the COVID-19 disease, has been the focus of concern around the world, setting records as a modern-day pandemic. The Centers for Disease Control has confirmed over 86 million cases in the world with over 1.9 million confirmed deaths as of January 5, 2021. SARS-CoV-2 continues to be a serious threat to global public health, leaving a distinct footprint in human history. Our research visualizes and contextualizes the mechanism of remdesivir, an antiviral nucleotide analog prodrug. Remdesivir targets the viral RNA-dependent RNA polymerase (RdRp) to inhibit replication of the virus. This process is facilitated by a subunit replication-and-transcription complex of three major nonstructural proteins (nsps): nsp7, nsp8, and nsp12. With our three-dimensional model (PDB ID 7BV2), we depict the mechanism of remdesivir inhibition of the copying of the viral RNA template through the central channel of RdRp by terminating chain elongation. When deciding the project, remdesivir showed promise as an inhibition drug for SARS-CoV-2, as it was effective for MERS-CoV. The National Institutes of Health and the World Health Organization have conflicting results about the effectiveness of remdesivir for treating COVID-19. Further comparison between the RdRp structures of SARS-CoV-2 and MERS-CoV may elucidate why remdesivir may be ineffective and could lead to better drug design. In our RNA model, the hydrogen bonds between the RNA primer strand and remdesivir are highlighted to show how remdesivir acts as a nucleotide to inhibit further viral replication. In the full RdRp complex, the hydrogen bonds between remdesivir and key residues of the nsp proteins are depicted. Our model illustrates the viral RdRp of SARS-CoV-2 and how remdesivir should impact viral RNA replication through inhibition.
The SARS-CoV-2 virus, which causes the COVID-19 disease, has been the focus of concern around the world, setting records as a modern-day pandemic. The Centers for Disease Control has confirmed over 86 million cases in the world with over 1.9 million confirmed deaths as of January 5, 2021. SARS-CoV-2 continues to be a serious threat to global public health, leaving a distinct footprint in human history. Our research visualizes and contextualizes the mechanism of remdesivir, an antiviral nucleotide analog prodrug. Remdesivir targets the viral RNA-dependent RNA polymerase (RdRp) to inhibit replication of the virus. This process is facilitated by a subunit replication-and-transcription complex of three major nonstructural proteins (nsps): nsp7, nsp8, and nsp12. With our three-dimensional model (PDB ID 7BV2), we depict the mechanism of remdesivir inhibition of the copying of the viral RNA template through the central channel of RdRp by terminating chain elongation. When deciding the project, remdesivir showed promise as an inhibition drug for SARS-CoV-2, as it was effective for MERS-CoV. The National Institutes of Health and the World Health Organization have conflicting results about the effectiveness of remdesivir for treating COVID-19. Further comparison between the RdRp structures of SARS-CoV-2 and MERS-CoV may elucidate why remdesivir may be ineffective and could lead to better drug design. In our RNA model, the hydrogen bonds between the RNA primer strand and remdesivir are highlighted to show how remdesivir acts as a nucleotide to inhibit further viral replication. In the full RdRp complex, the hydrogen bonds between remdesivir and key residues of the nsp proteins are depicted. Our model illustrates the viral RdRp of SARS-CoV-2 and how remdesivir should impact viral RNA replication through inhibition.
Hamline University (St. Paul MN)
Changes in SARS-CoV-2 Spike Protein Impacting Infectivity and Transmissibility
Team members: Patricia Hill, Lauren McDonald, Dean Young
Faculty advisor: Betsy Martinez-Vaz, Ph.D.
Project Overview
The Hamline CREST team is exploring evolutionary changes in the SARS-CoV-2 spike protein and the impact of these molecular variants on disease transmission and severity.
PDB IDs: 6zgg, 6xs6, 6vsb
Abstract
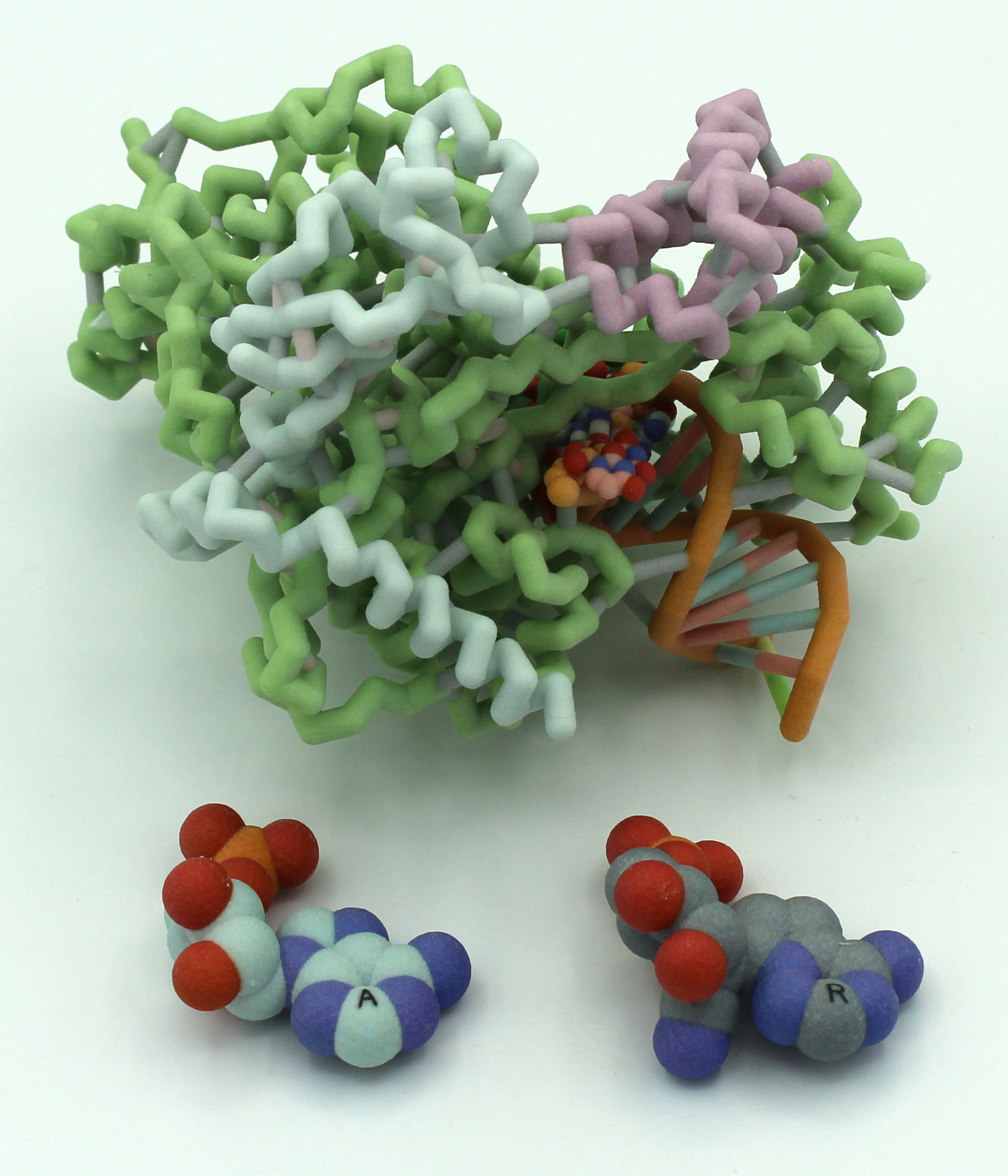
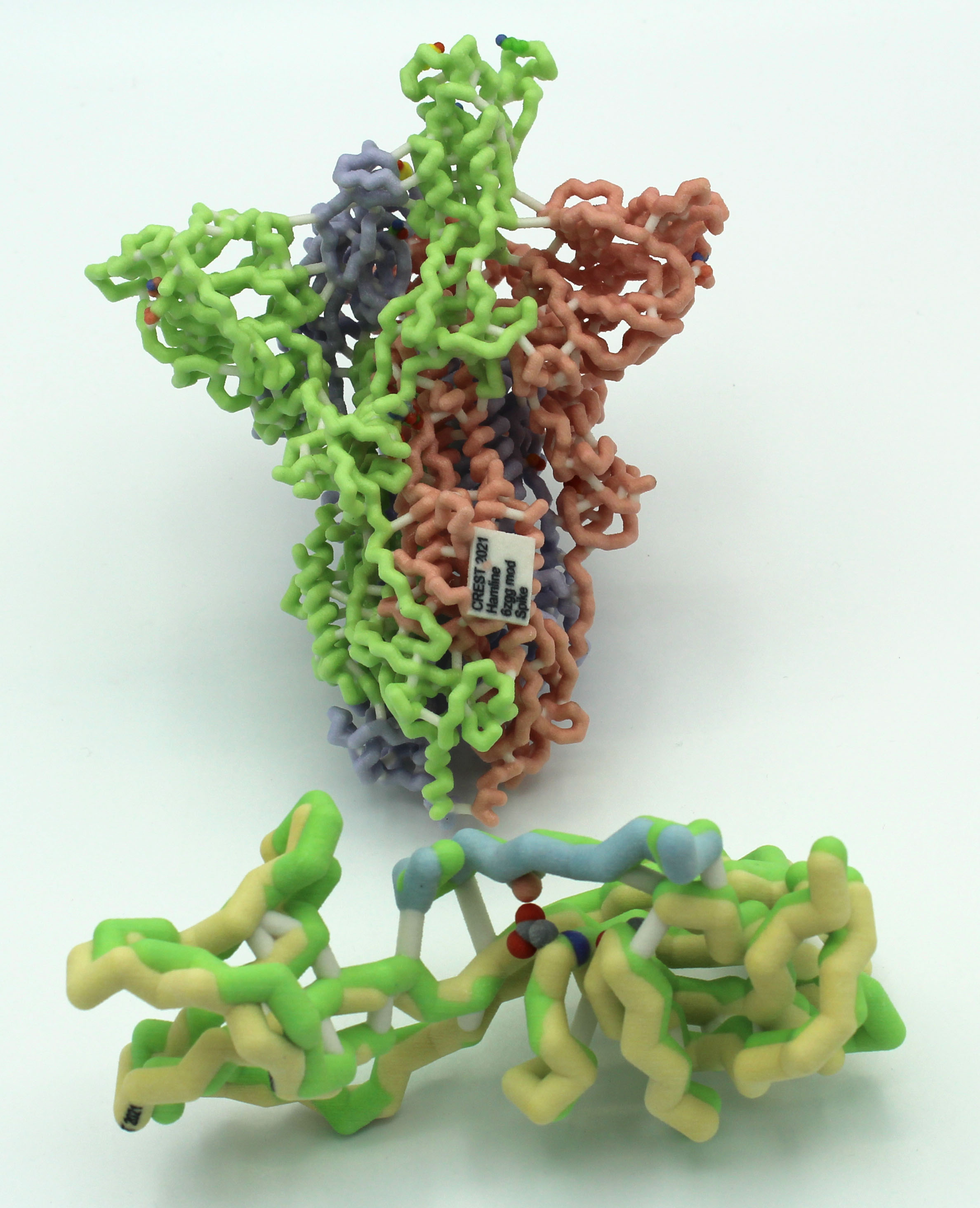 SARS-CoV-2 has become an increasingly dangerous virus infecting many people and resulting in over 2 million deaths in the first year of the pandemic. The spike glycoprotein plays an important role in the viral infection process by recognizing and binding to the ACE2 receptor in host’s cells. Due to the importance of this protein, current research efforts are focused on mutations that impact viral infectivity. SARS-CoV-2 viruses with a spike mutation at position 614 were recently shown to be more infectious than their wild-type ancestor. The D614G substitution eliminates a hydrogen bond with a threonine residue (T859) in an adjacent protein chain, allowing the spike protein to assume more readily an open conformation. Since the open conformation mediates attachment to the ACE2 receptor, the D614G mutant causes higher infectivity than the wild-type spike protein. The open conformation is further strengthened by shortening an intramolecular hydrogen bond between G614 and A647.
SARS-CoV-2 has become an increasingly dangerous virus infecting many people and resulting in over 2 million deaths in the first year of the pandemic. The spike glycoprotein plays an important role in the viral infection process by recognizing and binding to the ACE2 receptor in host’s cells. Due to the importance of this protein, current research efforts are focused on mutations that impact viral infectivity. SARS-CoV-2 viruses with a spike mutation at position 614 were recently shown to be more infectious than their wild-type ancestor. The D614G substitution eliminates a hydrogen bond with a threonine residue (T859) in an adjacent protein chain, allowing the spike protein to assume more readily an open conformation. Since the open conformation mediates attachment to the ACE2 receptor, the D614G mutant causes higher infectivity than the wild-type spike protein. The open conformation is further strengthened by shortening an intramolecular hydrogen bond between G614 and A647.
An active site box model was constructed to illustrate the biochemical interactions at the mutation sites. In this model, important amino acids Asp614, Ala647, and Thr859 are highlighted due to their role in hydrogen bonding and spike protein binding to the ACE2 receptor. A magnetic piece was created to show the effects of the aspartic acid to glycine mutation at position 614.
Lane College (Jackson TN)
SARS-CoV-2 RNA-dependent RNA Polymerase (RdRp) with bound Remdesivir
Team members: Valeanna Adams, Serena Barnhill, Ryan Billings, LeShaudria Brown, Michael Davis, Ntirenganyi Karamba, Tra’Mya Lauderdale
Faculty advisors: Candace Jones, Ph.D. and Melanie Van Stry, Ph.D.
Introductory Video
Project Overview
The Lane CREST team is exploring the interaction of Remdesivir with the SARS-CoV-2 RNA dependent RNA polymerase using pdb file 7bv2.
PDB ID: 7bv2
Abstract
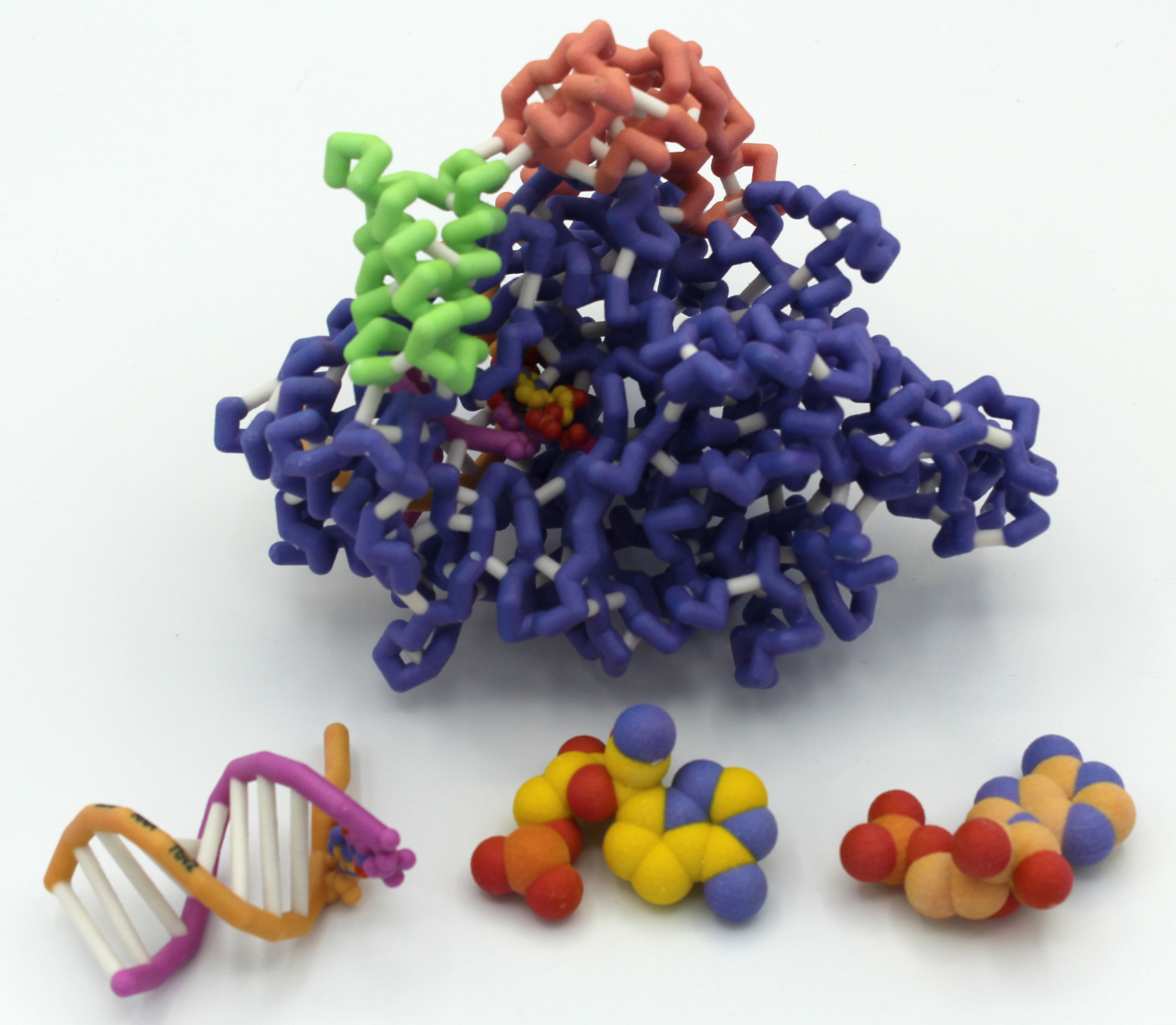 SARS-CoV-2 has become an increasingly dangerous virus infecting many people and resulting in over 2 million deaths in the first year of the pandemic. The spike glycoprotein plays an important role in the viral infection process by recognizing and binding to the ACE2 receptor in host’s cells. Due to the importance of this protein, current research efforts are focused on mutations that impact viral infectivity. SARS-CoV-2 viruses with a spike mutation at position 614 were recently shown to be more infectious than their wild-type ancestor. The D614G substitution eliminates a hydrogen bond with a threonine residue (T859) in an adjacent protein chain, allowing the spike protein to assume more readily an open conformation. Since the open conformation mediates attachment to the ACE2 receptor, the D614G mutant causes higher infectivity than the wild-type spike protein. The open conformation is further strengthened by shortening an intramolecular hydrogen bond between G614 and A647.
SARS-CoV-2 has become an increasingly dangerous virus infecting many people and resulting in over 2 million deaths in the first year of the pandemic. The spike glycoprotein plays an important role in the viral infection process by recognizing and binding to the ACE2 receptor in host’s cells. Due to the importance of this protein, current research efforts are focused on mutations that impact viral infectivity. SARS-CoV-2 viruses with a spike mutation at position 614 were recently shown to be more infectious than their wild-type ancestor. The D614G substitution eliminates a hydrogen bond with a threonine residue (T859) in an adjacent protein chain, allowing the spike protein to assume more readily an open conformation. Since the open conformation mediates attachment to the ACE2 receptor, the D614G mutant causes higher infectivity than the wild-type spike protein. The open conformation is further strengthened by shortening an intramolecular hydrogen bond between G614 and A647.
Milwaukee School of Engineering (Milwaukee WI) – Team 1
SARS-CoV-2 RNA-dependent RNA polymerase (RdRp) bound with Remdesivir
Team members: Evan Connelly, Avi Dutzi, Natalie Evans, Muskan Kanungo, Ryan Nickel, Amber Sabin, Keagan Schmidt
Faculty advisor: Anne Alexander, Ph.D.
Project Overview
The MSOE Remdesivir Team is interested in exploring how Remdesivir works to block viral infection. They are still in the planning stages of their project.
PDB ID: 7bv2
Abstract
 COVID-19 is a contagious disease caused by the SARS-CoV-2 strain of coronavirus, thought to be derived from bat coronaviruses due to the genetic similarities. Scientists began studying the effects of known antiviral medications on COVID-19 in hopes of quickly finding a suitable drug. One such medication was remdesivir, which was previously found to be somewhat successful in treating SARS and MERS coronaviruses in clinical trials. Although authorized for emergency treatment, it was deemed ineffective after studies found that remdesivir doesn’t affect the mortality rate of patients with COVID-19 in a statistically significant way.
COVID-19 is a contagious disease caused by the SARS-CoV-2 strain of coronavirus, thought to be derived from bat coronaviruses due to the genetic similarities. Scientists began studying the effects of known antiviral medications on COVID-19 in hopes of quickly finding a suitable drug. One such medication was remdesivir, which was previously found to be somewhat successful in treating SARS and MERS coronaviruses in clinical trials. Although authorized for emergency treatment, it was deemed ineffective after studies found that remdesivir doesn’t affect the mortality rate of patients with COVID-19 in a statistically significant way.
Remdesivir is a competitive inhibitor that resembles adenosine and works by binding to the RNA-dependent RNA polymerase (RdRp) that coronaviruses use to replicate their genomes. Remdesivir then causes an instability that terminates the RNA replication 3 base pairs upstream from the binding site, thus inhibiting the RdRp’s function. The RdRp of SARS-CoV-2 is made up of three non-structural proteins: NSP7, NSP8, and NSP12. NSP7 and NSP8 act as coenzymes to NSP12, allowing the conformational changes needed in the protein so that it can properly bind to the RNA bases. The RdRp also contains two magnesium ions in the active site that help stabilize the negative charge from the RNA backbone. Remdesivir itself has a few key differences from adenosine, the most prominent being a cyano group (CN) attached to C1’ of the ribose sugar. It is this group that is thought to cause the structural instability that terminates RNA replication.
Milwaukee School of Engineering (Milwaukee WI) – Team 2
Monoclonal Antibody S309 Binding to SARS COV-2 Spike Protein
Team members: Anna DeBruine, Alexa De La Sancha, Jude Ingham, Delila Payton, Krishna Persaud, Josephine Selvic, Behrgen Smith, Everen Wegner, Joshua Zieman
Faculty advisor: Anne Alexander, Ph.D.
Project Overview
The MSOE Antibody Team is exploring the interactions of the human monoclonal antibody S309 with the receptor binding domain of the spike protein in both open (6wpt.pdb) and closed (6wps) forms. They are exploring molecular interactions at the binding site based on work by Pinto et al., 2020.
PDB ID: 6wps
Abstract
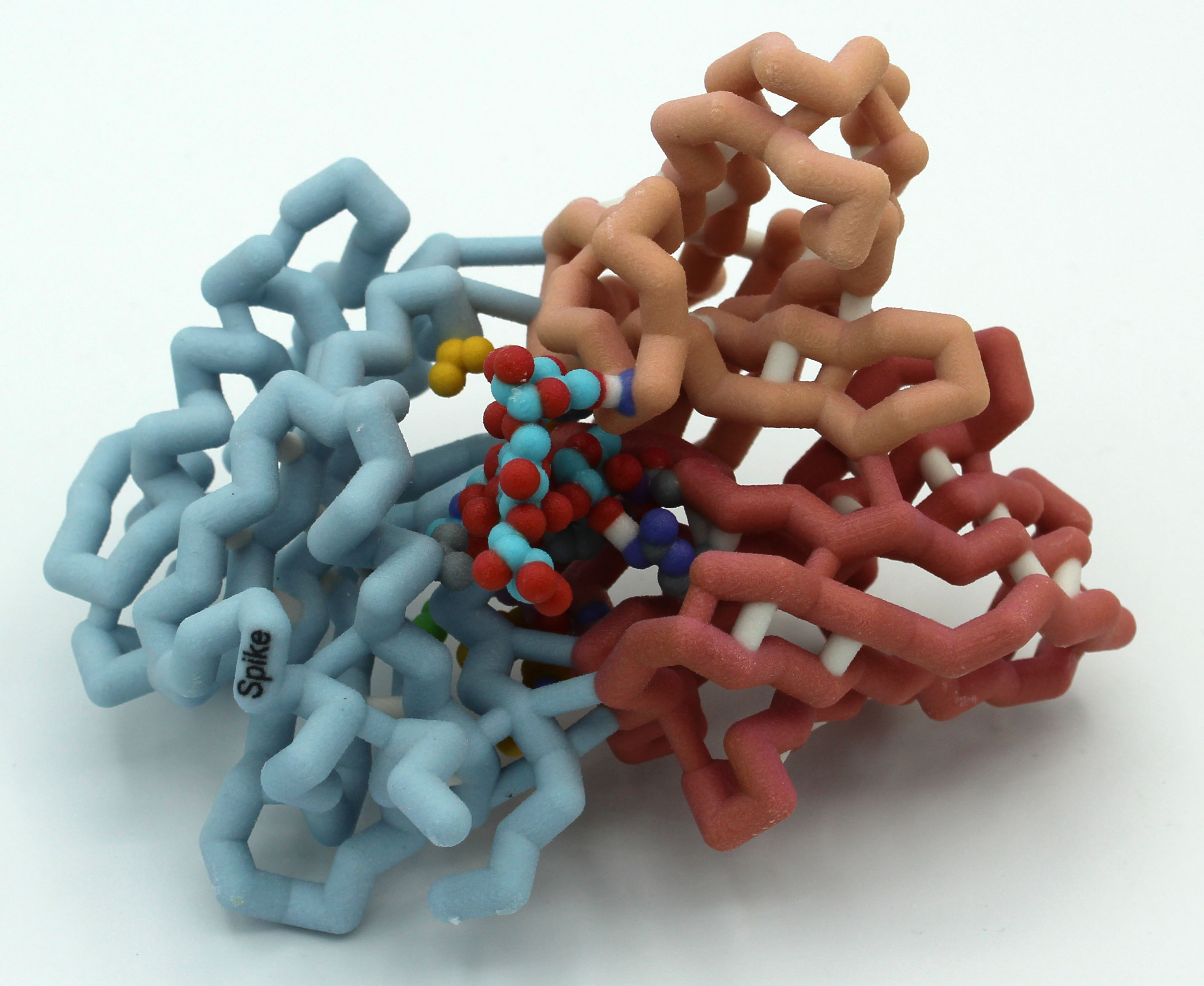 S309 is a monoclonal antibody (mAb) that potentially neutralizes the SARS-CoV-2 virus, a respiratory coronavirus, causative agent of the COVID-19 pandemic. mAbs differ from vaccines in that they are a treatment and do not activate antibody production or confer long-term immunity. Due to the conserved structures across SARS-CoV variants, an understanding of protein-mAb interactions and subsequent impacts on disease offer a pathway to developing future treatments for novel vaccine-resistant variants. S309 was designed from an antibody that inhibits the SARS-CoV spike protein, a transmembrane protein that mediates entry into a host cell. S309 was designed to be cross-reactive for SARS-CoV-2 as well. The S309 mAb binds noncompetitively to a conserved glycan (sugar) on the spike protein as opposed to the ACE-2 receptor binding motif. Along with the glycan, S309 recognizes several amino acid residues within the epitope, binding and subsequently neutralizing the virus, although the specific conformational change that leads to neutralization is unknown. Using Jmol, we identified significant intramolecular interactions by isolating amino acid residues involved in the binding of the antibody to the spike protein. Significant interactions were defined as being within a proximity of five Å to other binding residues and binding type was determined by the strongest attraction. Of the major interactions, two hydrophobic pockets, one ionic bond, two hydrogen bonds and two dipole-dipole interactions were considered integral components of the S309-spike protein binding, along with three London Dispersion Forces interactions that position the glycan.
S309 is a monoclonal antibody (mAb) that potentially neutralizes the SARS-CoV-2 virus, a respiratory coronavirus, causative agent of the COVID-19 pandemic. mAbs differ from vaccines in that they are a treatment and do not activate antibody production or confer long-term immunity. Due to the conserved structures across SARS-CoV variants, an understanding of protein-mAb interactions and subsequent impacts on disease offer a pathway to developing future treatments for novel vaccine-resistant variants. S309 was designed from an antibody that inhibits the SARS-CoV spike protein, a transmembrane protein that mediates entry into a host cell. S309 was designed to be cross-reactive for SARS-CoV-2 as well. The S309 mAb binds noncompetitively to a conserved glycan (sugar) on the spike protein as opposed to the ACE-2 receptor binding motif. Along with the glycan, S309 recognizes several amino acid residues within the epitope, binding and subsequently neutralizing the virus, although the specific conformational change that leads to neutralization is unknown. Using Jmol, we identified significant intramolecular interactions by isolating amino acid residues involved in the binding of the antibody to the spike protein. Significant interactions were defined as being within a proximity of five Å to other binding residues and binding type was determined by the strongest attraction. Of the major interactions, two hydrophobic pockets, one ionic bond, two hydrogen bonds and two dipole-dipole interactions were considered integral components of the S309-spike protein binding, along with three London Dispersion Forces interactions that position the glycan.
Nova Southeastern University (Fort Lauderdale FL)
Minor changes with large implications: Modeling amino acid mutations in COVID-19 monoclonal antibodies (80R and 362) towards the design of more universal antibodies
Team members: Feza Abbas, Lyla Abbas, Carolina Alzamora, Carol Manikkuttiyil, Christo Manikkuttiyil, Matthew Hunt, Pujita Julakanti, Sanjana Likki
Faculty advisors: Emily Schmitt, Ph.D. and Arthur Sikora, Ph.D.
Introductory Video
Project Overview
The Nova Team has been exploring the structure of two antibodies to SARS viruses. The 80R antibody only binds to SARS-CoV-1 (2ghw.pdb) but not SARS-CoV-2. The Schiffer lab and their collaborators used computational techniques to create MAb362, an antibody that binds to BOTH SARS-CoV-1 and SARS-CoV-2.(Ejemel et al., 2020). The team is working with Shurong Hou, Ph.D., a post-doc in the Schiffer lab and coauthor of the paper, to build models of the spike protein receptor binding domain (to which the antibodies attach) of both viruses, as well as the two antibodies. They will use the models to better understand the interactions of the protein and antibodies. They are hoping to design their own antibody based on what they learn in the project.
PDB IDs: 3ghw and unpublished structures
Abstract
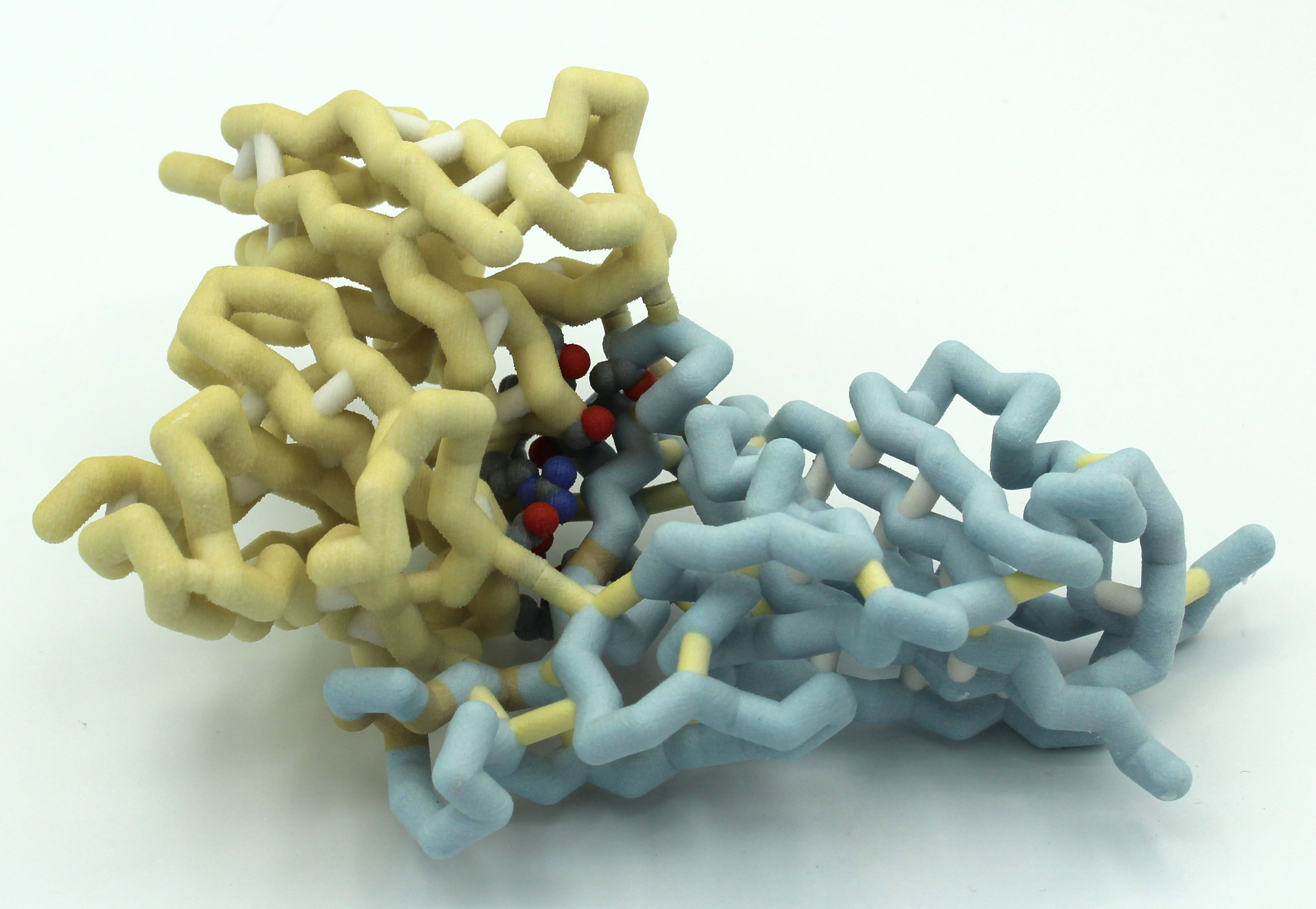 The SARS-CoV2 virus that caused the COVID-19 pandemic has impacted nearly every person on Earth and caused over 1.3 million deaths. The current anti-SARS-CoV monoclonal antibodies (MAb) 80R and 362 bind to epitopes on the spike protein receptor-binding domain (RBD) to neutralize the virus (Walls et al., 2020). To better explain this process, the NOVA CREST team modeled and compared the 80R antibody that binds to SARS-CoV1 and the MAb362 antibody that can bind to both SARS-CoV1 and SARS-CoV2. Models were designed to show the 80R and 362 antibodies binding to the RBD of the two viral spike proteins. By studying the point mutations made between the antibodies 80R and 362, we modeled a potentially more universal antibody that may be
The SARS-CoV2 virus that caused the COVID-19 pandemic has impacted nearly every person on Earth and caused over 1.3 million deaths. The current anti-SARS-CoV monoclonal antibodies (MAb) 80R and 362 bind to epitopes on the spike protein receptor-binding domain (RBD) to neutralize the virus (Walls et al., 2020). To better explain this process, the NOVA CREST team modeled and compared the 80R antibody that binds to SARS-CoV1 and the MAb362 antibody that can bind to both SARS-CoV1 and SARS-CoV2. Models were designed to show the 80R and 362 antibodies binding to the RBD of the two viral spike proteins. By studying the point mutations made between the antibodies 80R and 362, we modeled a potentially more universal antibody that may be 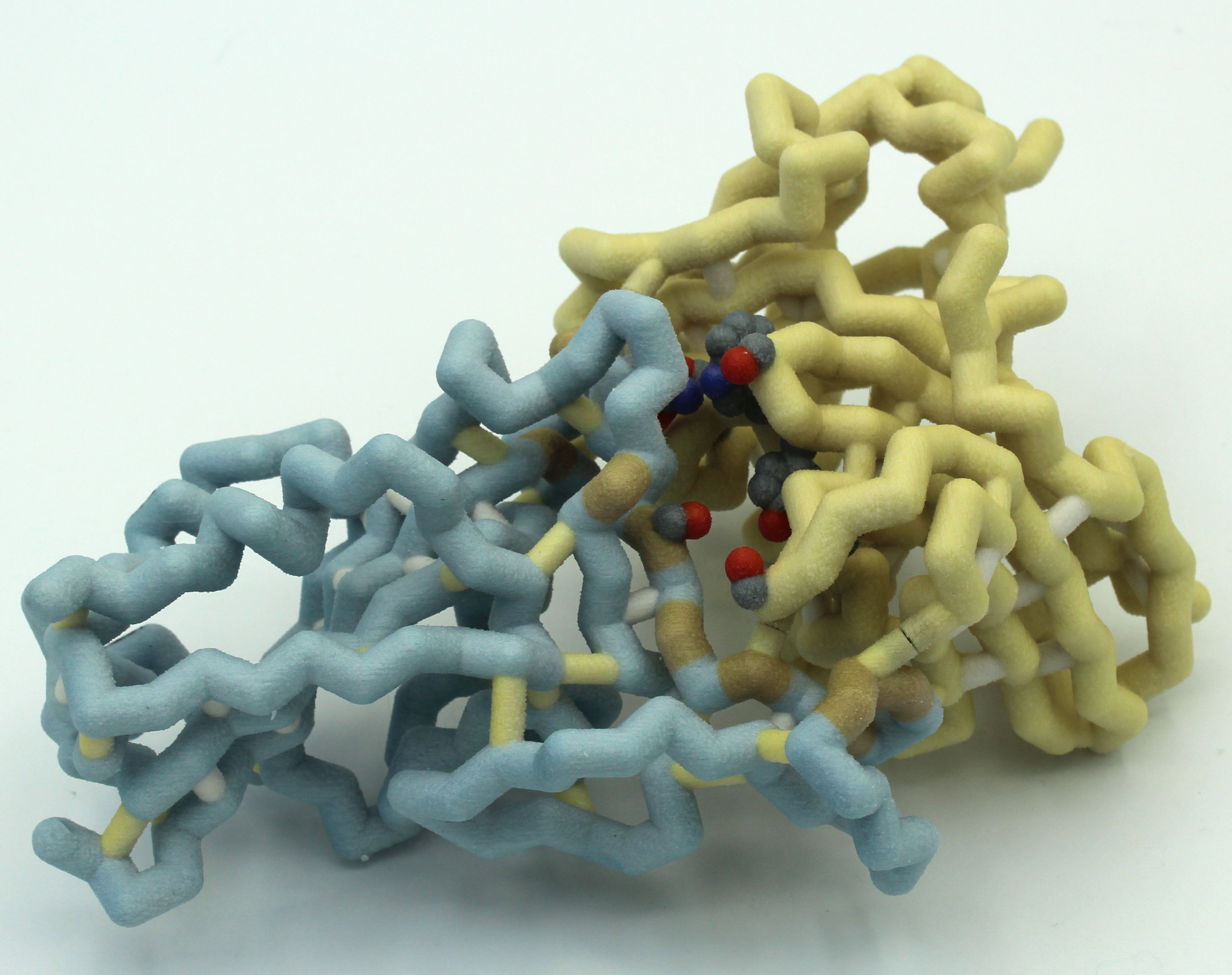 able to bind to future mutations in the SARS spike protein. The cross-neutralizing MAb362 antibody also binds to the spike protein to block ACE2 receptor binding, neutralizing SARS-CoV2. At the binding interface, MAb362 mutations trend smaller and less polar including, Arg149Ser, Asn151Ser, Asp170Gly, and Trp213Ser. Due to the trend of smaller amino acids appearing in the MAb362 binding interface, we hypothesize that more space in this area could allow antibodies to be more resistant to COVID-19 spike protein structure variations. NSU1 is modeled based on Mab362 with
able to bind to future mutations in the SARS spike protein. The cross-neutralizing MAb362 antibody also binds to the spike protein to block ACE2 receptor binding, neutralizing SARS-CoV2. At the binding interface, MAb362 mutations trend smaller and less polar including, Arg149Ser, Asn151Ser, Asp170Gly, and Trp213Ser. Due to the trend of smaller amino acids appearing in the MAb362 binding interface, we hypothesize that more space in this area could allow antibodies to be more resistant to COVID-19 spike protein structure variations. NSU1 is modeled based on Mab362 with 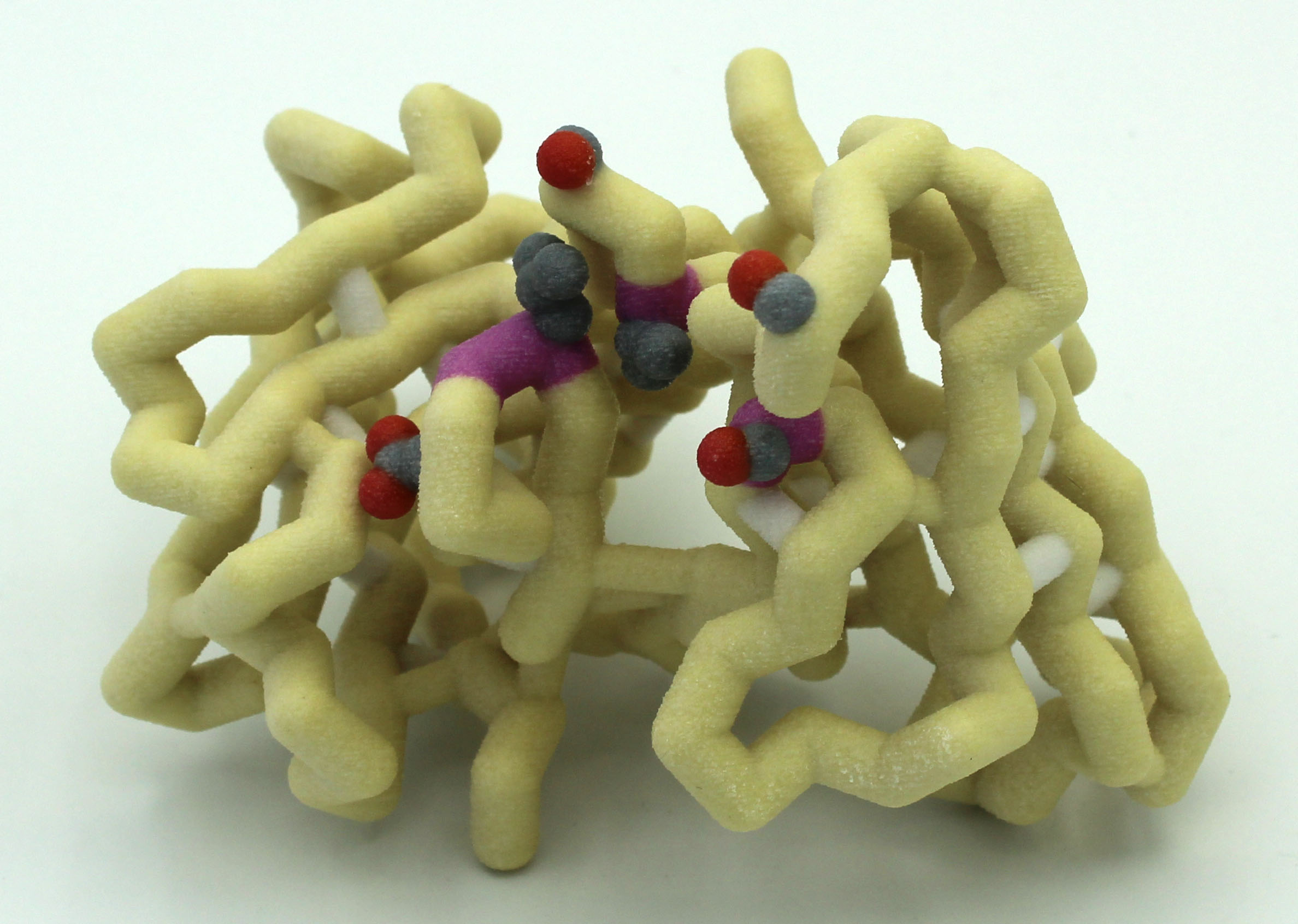 the following four additional mutations: Asp103Gly, Trp104Leu, Gly170Ser, and Arg211Val based on mutation trends presented by Mirsky et al., 2015. All of these except for Gly170 are mutations that decrease in size and polarity. Position 170 is Asp on the 80R structure and thus a mutation to Ser is expected to maintain this trend. Due to this increased binding site size, NSU1 is predicted to be more resistant to future COVID-19 spike protein mutations.
the following four additional mutations: Asp103Gly, Trp104Leu, Gly170Ser, and Arg211Val based on mutation trends presented by Mirsky et al., 2015. All of these except for Gly170 are mutations that decrease in size and polarity. Position 170 is Asp on the 80R structure and thus a mutation to Ser is expected to maintain this trend. Due to this increased binding site size, NSU1 is predicted to be more resistant to future COVID-19 spike protein mutations.
Virtual Poster Presentations at ASBMB Annual Meeting, April 27-30, 2021
SARS-CoV-2 ORF8 Accessory Protein Dimerization Domains and Protein-Protein Host Interaction
Allison Robinson*, Ryan Peterson, Leeann Stearns, Ryan Vazquetelles, Elizabeth Wiles, Bailey Hart, Karen Guzman
Campbell University, Buies Creek NC
3D Physical Models of the SARS-CoV-2 Spike Protein: Exploring the Impact of the D614G Mutation on Viral Infectivity
Lauren McDonald*, Dean Young, Trisha Hill
Hamline University, St. Paul MN
Exploring the Mechanisms of Antiviral Therapeutics: A 3D Model of SARS-CoV-2 RdRp in Complex with Remdesivir
Michael Davis*, Valeanna Adams, Serena Barnhill, Ryan Billings, LeShaundria Brown, Tra’Mya Lauderdale, Ntirenganyi Karamba, Melanie Van Stry, Candace Jones Carter
Lane College, Jackson TN
Minor Changes with Large Implications: Modeling Amino Acid Mutations in SARS-CoV Monoclonal Antibodies (80R and 362) Towards the
Design of More Universal Antibodies
Carol Manikkuttiyil*, Feza Abbas, Lyla Abbas, Carolina Alzamora, Matthew Hunt, Pujita Julakanti, Sanjana Likki, Christo Manikkuttiyil, Emily Schmitt
Lavin, Arthur Sikora
Nova Southeastern University, Fort Lauderdale FL
Virtual Poster Presentations at PDB50, May 4-5, 2021
SARS-CoV-2 ORF8 Accessory Protein Dimerization Domains and Protein-Protein Host Interaction
Elizabeth Wiles*, Ryan Peterson, Allison Robinson, Leeann Stearns, Ryan Vazquetelles, Bailey Hart, Karen Guzman
Campbell University, Buies Creek NC
3D Physical Models of the SARS-CoV-2 Spike Protein: Investigating Mutations Associated with High Infectivity and Transmissibility.
Dean Young*, Lauren McDonald, Trisha Hil
Hamline University, St. Paul MN
Exploring the Mechanisms of Antiviral Therapeutics: A 3D Model of SARS-CoV-2 RdRp in Complex with Remdesivir
LeShaundria Brown*, Michael Davis, Valeanna Adams, Serena Barnhill, Ryan Billings, Tra’Mya Lauderdale, Ntirenganyi Karamba, Melanie Van Stry, Candace Jones Carter
Lane College, Jackson TN
Remdesivir and the RNA-dependent RNA polymerase of SARS-CoV-2
Muskan Kanungo, Amber Sabin, Evan Connelly, Natalie Evans, Ryan Nickel, and Keagan Schmidt
Milwaukee School of Engineering, Milwaukee WI
Using the protein databank to visualize the effect of minor amino acid differences on the function of SARS-CoV monoclonal antibodies
Sanjana Likki*, Feza Abbas, Lyla Abbas, Carolina Alzamora, Matthew Hunt, Pujita Julakanti, Carol Mannikkuttiyil, Christo Mannikkuttiyil, Emily Schmitt Lavin and Arthur Sikora
Nova Southeastern University, Fort Lauderdale FL
*Lead presenter
Questions about the CREST Program? Contact Margaret Franzen at franzen@msoe.edu or 414-277-2806. We look forward to hearing from you!
