Molecular Pharmacology Poster Session – April 4, 2013
The Molecular Pharmacology students at Concordia University Wisconsin School of Pharmacy working with Dr. Dan Sem explored the molecular mechanism of drug action. Teams of students met with preceptors (pharmacist mentors) to discuss interesting clinical cases in which a prescribed drug resulted in either unwanted side effects or drug-drug interactions. Each team then explored one of these drug stories, developing a case study, historical background on drug development and the mechanism of drug action. The project involved writing a group paper, creating a 3D model of the protein using Jmol and 3D printing technology, and presenting a poster at the poster session.
Protein models depict the drug docked with the target protein; additional models were subsequently generated, showing a close up of the active site of the protein with the drug docked using magnets. These models are available for loan through the MSOE Model Lending Library. Contact (crestprogram@gmail.com) for information about borrowing this collection.
Xanthine Oxidase with Bound Zyloprim (Allopurinol)
Poster Development Students:

- Biniam Berhane
- Jacob Bognar
- Kaitlyn Garr
- Dylan Jones
- Eric Newenhouse
- Sarah Seward
Model Development Students:
- Tyler Frisque
- Natalie Koenig
- Cameron Patterson
Structure:
- Protein: xanthine oxidase
- Drug: allopurinol (Zyloprim)
- PDB file: 3bdj.pdb
Presentation:
Abstract:

Gout is a painful arthritic condition characterized by uric acid levels in the blood, resulting in deposits in joints, tendons, and tissues. Although the exact cause of gout is unknown, diet and genetic have been linked to its occurrence. Xanthine oxidase, an enzyme that aids in the metabolism of purine bases, plays a major role in this process. Oxidation of purines by xanthine oxidase produces uric acid, which may accumulate over time, causing pain and inflammation in the joints. Treatment of gout typically involves xanthine oxidase inhibitors, such as allopurinol. Although allopurinol is metabolized quickly, its active metabolite, oxypurinol, remains present and further inhibits the actions of xanthine oxidase. Rheumatoid arthritis is an autoimmune disease involving pain and inflammation in joints. The cause of rheumatoid arthritis is unclear, but may have a genetic component. Immunosuppressive medications such as azathioprine are used in treatment regimens to suppress the immune system’s attack on healthy tissue. Azathioprine, a prodrug, requires conversion into its active form, 6-mercaptopurine, which is inactivated via oxidation by xanthine oxidase. A drug interaction occurs between allopurinol and azathioprine due to their requirements for xanthine oxidase. Because the allopurinol metabolite is a competitive inhibitor of xanthine oxidase, levels of 6-mercaptopurine may dramatically increase. Proper precaution should be used when taking these medications concomitantly. A dose reduction in azathioprine may be required to limit toxic levels of 6-mercaptopurine.
μ-Opioid Receptor with Bound β-FNA (Opiate)
Poster Development Students:

- Nathan Bahr
- Michael Gremban
- Kurt LaRock
- Alyssa Meixelsperger
- Samantha Stancato
Model Development Students:
- Andrea Brandt
- Bradley Kendziora
- Robert Schumacher
- Alexandru Ulici
Structure:
- Protein: μ-opioid receptor
- Drug: generic opiate (β-FNA)
- PDB file: 4dkl.pdb
Presentation:
Abstract:

Opiates are natural products from the Papaver somniferum poppy plant. Opioid analgesics, such as oxycodone and hydromorphone, are opiate derivatives that bind to opioid receptors (OR) and have a long history of use for pain management. ORs are found in the periphery, spinal cord, and cerebral cortex. Three types of OR subtypes exist: mu, kappa, and delta. Clinically used opioid analgesics act on all OR subtypes, but have highest affinity for the mu-opioid receptor (MOR) as it is the major target for pain relief. MOR activation by opiates, endogenous ligands, and antagonists induces euphoria, analgesia, and opioid toxicity reversal. MOR agonists can have undesirable effects, such as respiratory depression, which can be reversed with an opioid antagonist, such as naloxone. Naloxone exhibits a competitive reversal effect on the MOR. The structural similarity between naloxone and opioid agonists shows how small changes in the pharmacophore can have significant changes in the action of drugs. Binding of opioid agonists and antagonists to the MOR is not fully understood. However, mutation studies have shown that there are necessary ionic bonds formed between charged amino acids in the receptor and the ligand. Action of ligand binding at the MOR is difficult to capture on a crystal structure because of the transient nature of the bonds. Beta-Funaltrexamine (β-FNA) is a mimic for a bound ligand such as naloxone or oxycodone and irreversibly binds to the Mu-opioid receptor. β-FNA enables visualization of interactions between a ligand and amino acids of the MOR transmembrane domains involved in binding.
G-Coupled β1-Adrenergic Receptor with Bound Proventil (Albuterol Sulfate)
Poster Development Students:

- Rostian Akpor-Mensah
- Laura Clark
- Brooke Koenig
- Emily Komp
- Ashley Voigt
- Jennifer Watzig
Model Development Students:
- Jacob Kachelmeier
- Adam Naker
- Staci Prise
Structure:
- Protein: G-coupled β1-adrenergic receptor
- Drug: albuterol sulfate (Proventil)
- PDB file: 2y04.pdb
Presentation:
Abstract:

Upon stimulation of the β2-adrenergic receptors in the lungs through inhalation of the medication, (R)-albuterol exhibits a sympathomimetic response of relaxing smooth muscles and relieving bronchospasm. (R)-albuterol can also stimulate the β1-adrenergic receptors in the heart, resulting in an increased rate and force of myocardial contractions. This off-target effect of the asthma medication has been known to cause unwanted side effects such as racing heart rate and jitters. The (R)-albuterol molecule has three domains that interact with amino acids in the binding site of the β2-adrenergic receptor. These domains consist of a catechol ring, a hydroxyl group, and an amine. The secondary amine forms a hydrogen bond with Asn329 and with Asp121, acting as the hydrogen donor in both interactions. The hydroxyl group of the carbon chain interacts with Asp121 as a hydrogen donor and with Asn329 as a hydrogen acceptor. The catechol ring interacts with three different amino acids of the β2-adrenergic receptor. One of these interactions is theπ-πinteraction between the catechol ring and Phe307 of the receptor. Van der Waals interactions with the catechol ring and the neighboring chains in the receptor enhance the binding of (R)-albuterol. The para-hydroxyl group on the catechol ring forms a hydrogen bond with Ser211 ((R)-albuterol acting as the hydrogen donor), while the meta-hydroxymethyl of the catechol ring forms a hydrogen bond with the slightly negative charge of Asn310 ((R)-albuterol acting as the hydrogen donor).
cGMP-Specific Phosphodiesterase type 5 (PDE-5) with Bound Viagra (Sildenafil)
Poster Development Students:

- Kevin Frank
- Chris Johnson
- Kristen Jones
- Will Mahoney
- Jade Senger
- Amy Yang
Model Development Students:
- Adam Richter
- Austin Ross
Structure:
- Protein: cGMP-specific phosphodiesterase type 5 (PDE-5)
- Drug: sildenafil (Viagra)
- PDB file: 2h42.pdb
Presentation:
Abstract:

Roughly 35% of American men aged 40-70 years are affected by erectile dysfunction (ED); this may be an underestimate due to the unwillingness patients may have in discussing this condition with their doctors. As the number of aging adults increases in the US, the incidence of ED is expected to rise. Many factors may contribute to or cause ED; these factors fall into three categories: organic (neurogenic, hormonal, or drug-induced), psychogenic, or mixed organic and psychogenic. Oral medications are first line therapy for ED. One option is a PDE-5 inhibitor such as sildenafil (Viagra). Sexual stimulation increases in cGMP and nitric oxide (NO) release. This causes smooth muscle relaxation and an influx of blood into the tissue of the penis causing an erection. PDE-5 is an enzyme whose role is to break down cGMP. Sildenafil blocks cGMP breakdown by binding to the active site of PDE-5 where cGMP normally binds. One amino acid in the binding pocket, glutamine (green), stabilizes the negative and positive charges on the drug molecule. The other amino acids illustrated (purple) in the pocket interact with the non-charged portion of the drug via Van der Waals interactions. Interactions of the drug with the binding pocket can vary, due to the flexibility of the H loop (red). Depending on the position of the H-loop, some amino acids may or may not be interacting with the drug and therefore may change the binding affinity.
Thyroid Hormone Receptor with Bound Synthroid (Levothyroxine)
Poster Development Students:

- Ardalan Abtahi
- Kelly DeCanio
- Kayla Jacoby
- James Mahoney
- Thomas Nowak
Model Development Students:
- David Grandinetti
- Michael Holyoke
Structure:
- Protein: thyroid hormone receptor
- Drug: levothyroxine (Synthroid)
- PDB file: 1y0x.pdb
Presentation:
Abstract:
<
Thyroid Hormone Receptor (TR) is responsible for a wide array of physiologic functions including metabolism, cognition, and heart rate. Therefore, in the case of deficient endogenous hormones, it is imperative to replace them to maintain proper thyroid function. A deficiency of thyroid hormones (T3 and T4) can lead to heart disease, joint pain, obesity, and infertility. Levothyroxine is a synthetic form of T4 and it binds TR with the same affinity. It is also mediated by the same binding contacts as T4. There are hydrogen bonds between the drug and both Arg282 and His435. Notably, T4 binds with less affinity than T3, so drug therapy replaces only a portion of the natural mechanism. Additionally, albumin and other proteins compete for levothyroxine, though these things are all accounted for in dosing ranges. Levothyroxine is thought to potentiate the effects of warfarin, calling for a dose reduction when the drugs are used concomitantly. To date, no controlled studies have been performed, so more research in this area may be needed. This commonly used medication has far-reaching effects, and its ability to replace endogenous hormones is important for those with hypothyroidism.
G-Coupled β1-Adrenergic Receptor with Bound Coreg (Carvedilol)
Poster Development Students:

- Carmon Breitlow
- Kelly Fausek
- Mickey Hart
- Sarah Hoerner
- Birgitta Monson
Model Development Students:
- Melissa Duoss
- Alana Everson
- Landon Kortman
Structure:
- Protein:G-coupled β1-adrengergic receptor
- Drug:carevedilol (Coreg)
- PDB file: 4amj.pdb
Presentation:
Abstract:

Norepinephrine is a catecholamine which acts as both a hormone and a neurotransmitter involved in the sympathetic nervous system. An increase in norepinephrine levels can have several effects on the heart and vasculature in the body. In the heart, norepinephrine binds to the G-coupled protein β1-receptor, causing an increased heart rate. The current protocol for patients with heart failure is to place them on a beta-blocker, such as carvedilol, to block norepinephrine from reaching the receptor. Carvedilol (Coreg) is an adrenergic antagonist with α1, β1, and β2 blocking activity. Carvedilol is administered as a racemic mixture where the S(-) enantiomer is both an α and nonselective β-blocker, whereas the R(+) enantiomer is an α1-blocker. When bound to the β1-receptor, carvedilol binds to three amino acids found in the receptor (ser211, asn329 and asp121). Prevention of norepinephrine binding to the β1-receptor slows down the heart rate in heart failure patients allowing the ventricles to fill more adequately with blood. This allows the heart to pump better by ejecting more blood into the vasculature. While this is one recommended therapy for heart failure patients, a variety of medications are required to improve symptoms.
α4β2 Nicotinic Acetylcholine Receptor with Bound Chantix (Varenicline)
Poster Development Students:

- Karena Creten
- Elizabeth Everett
- Sirr Grice
- Margaret Roesser
Model Development Students:
- Joseph Holewa
- Veronica Juarez
- Thomas Puerzer
- Jacob Sutherland
Structure:
- Protein: α4β2 nicotinic acetylcholine receptor
- Drug:varenicline (Chantix)
- PDB file: 4afg.pdb
Presentation:
Abstract:
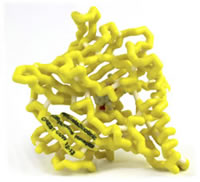
Tobacco dependence is the most common preventable cause of death in the United States with 443,000 attributed deaths and related costs amounting to an estimated $193 billion annually. Nicotine replacement therapy has been the standard of care for many years but shows mixed efficacy in getting smokers to quit. The addictive mechanism of nicotine is its mediation of synaptic neurotransmission within the body by binding to the α4β2 subtype of nicotinic acetylcholine receptors (nAChRs). This binding triggers the release of dopamine in the mesolimbic dopamine “reward” system. As the concentration of nicotine declines, the dopamine levels subside which signals the urge to smoke again. Varenicline (Chantix) is a partial agonist of the α4β2 subunits of nAChRs. Structurally, varenicline is made up of a benzazepine molecule with a pyrazine ring attached to the benzene ring. The pyrazine ring and the azepine ring of the molecule account for the major binding interactions within the nAChR binding site. As a partial agonist to α4β2 nAChR, varenicline blocks the binding of nicotine while still producing a dopaminergic release. This mechanism allows the patient to continue to smoke during therapy without receiving the acute dopaminergic release “reward” for smoking while still achieving some dopamine release to lessen withdrawal symptoms. The ability for the patient to continue the habit of smoking is an important benefit, as many quitting attempts fail because patients cannot break the behavioral aspect of their smoking addiction.
Viral Thymidine Kinase with Bound Zovirax (Acyclovir)
Poster Development Students:

- Caitlyn King
- James Le
- Marena Muelbl
- Tabitha Senfleben
Model Development Students:
- Christian Beck
- Dana Darnauer
- Marie Thomas
Structure:
- Protein:viral thymidine kinase
- Drug:acyclovir (Zovirax)
- PDB file: 2ki5.pdb
Presentation:
Abstract:

Acyclovir is an antiviral pro-drug that works by inhibiting viral replication. It is primarily used to treat and prevent two common Herpes simplex viruses: HSV-1 and HSV-2. Herpes simplex is not a curable disease state because it has a latent phase which cannot currently be targeted by drug therapy. During latency, the latency-associated transcript (LAT) is expressed which causes recurrent cases of HSV-1. Acyclovir is a guanosine nucleoside analog that requires phosphorylation in three distinct steps before it becomes active and inhibits the HSV DNA polymerase. Acyclovir is first phosphorylated by virus-encoded thymidine kinase (vTK) and then twice by host cellular thymidylate kinases. The final acyclovir triphosphate is the active competitor that inhibits the viral DNA polymerase and prevents replication. It does this by competing with 2-deoxyguanosine triphosphate (dGTP) as a substrate for viral DNA polymerase. This halt in DNA replication stops the proliferation of the virus and alleviates the symptoms in affected patients. Acyclovir interacts with several different amino acid side chains in the vTK active site via hydrogen bonding and Pi-Pi stacking.These bonds keep the nucleoside analogue within the binding pocket. Most acyclovir resistance is due to additions or deletions of nucleotides in the genes coding for vTK, which can cause a frameshift mutation resulting in amino acid substitutions that decrease acyclovir phosphorylation activity.
Cytochrome P450 2B6 (CYP450 2B6) with Bound Norvasc (Amlodipine)
Poster Development Students:

- Dewaa Ali
- Craig Bishop
- Kelly Cobb
- Kayla Deitte
- Brittany Jensen
- Michelle Meunier
Model Development Students:
- Katie Kotz
- Jason Slaasted
Structure:
- Protein:cytochrome P450 2B6 (CYP450 2B6)
- Drug:amlodipine (Norvasc)
- PDB file: 3ua5.pdb
Presentation:
Abstract:
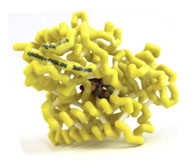
Amlodipine, methyl ethyl-(2-aminoethoxymethyl)-4-(2-chlorophenyl)-6-methyl-1,4-dihydropyridine-3,5-dicarboxylate, is a Calcium Channel Antagonist (CCA). CCAs bind calcium channels located on cardiac and smooth muscle, preventing the influx of calcium into the cell. This inhibition causes vasodilation and a decrease in blood pressure. CCAs are primarily metabolized into inactive metabolites through hepatic CYP450 isoenzymes. Amlodipine is metabolized by CYP450 3A4 and recently shown to interact with CYP450 2B6. The highest affinity pose of amlodipine in 2B6 shows a strong bond between the 2-aminoethoxymethyl nitrogen of amlodipine and the heme iron within 2B6. This interaction is important in aligning amlodipine in the active site of 2B6. In the active site a hydrogen bond is formed between the ethoxy oxygen of amlodipine and the hydroxyl of Thr302 in 2B6. Other amino acids (Phe297, Ala298, Leu363 and Val367) are believed to have Van der Waals interactions which further stabilize amlodipine in the active site of 2B6.Amlodipine’s interaction with 2B6 is important because many medications also interact with 2B6. Azoles, a group of antifungal medications, are particularly important because they are strong inhibitors of 2B6. When 2B6 is inhibited, Amlodipine is metabolized more slowly; and if the dose is not changed the patient can become hypotensive.
Dihydrofolate Reductase with Bound Trexall (Methotrexate)
Poster Development Students:

- Catherine Avis
- Tracy Drott
- Ashley Feltes
- Courtney Maust
- Benjamin Plass
Model Development Students:
- Brain Danihlik
- Kelsey Eckert
- Brigette Yoder
Structure:
- Protein:dihydrofolate reductase
- Drug:methotrexate (Trexall)
- PDB file: 1mvt.pdb
Presentation:
Abstract:
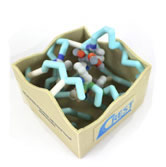
Methotrexate is an antimetabolite drug that is used in the treatment of rheumatoid arthritis. Methotrexate is one of the most potent inhibitors of dihydrofolate reductase (DHFR), which is needed for DNA and RNA synthesis. It has been thought that by preventing synthesis of purines and pyrimidines needed for DNA and RNA synthesis through the inhibition of DHFR, proliferation of quickly dividing lymphocytes and other cells that cause synovial inflammation can be prevented. Thus, patients could receive relief from symptoms of rheumatoid arthritis. Methotrexate is known as a classic DHFR inhibitor because it contains the same three groups as folic acid which are a pteridine moiety, PABA, and glutamic acid. The main difference is that methotrexate has a 2,4-diaminopyrimidine unit replacing the OH group with an amino group at the C-4 site. This group makes methotrexate more basic than folic acid because it contains an electron releasing amino group that is conjugated with the basic guanidine. This is in comparison to folic acid, which has an electron withdrawing carbonyl. This new addition of the 4-amino group makes methotrexate bind about 10³ times stronger than folate to the DHFR.