Marian University (Fond Du Lac WI)
Structure, Function and Mechanism of Action for Shiga Toxin
Team members: J. Brum, R. Panelo, P. Smith, A. Speck and T. Yang
Faculty advisor: Susan Bornstein-Forst, Ph.D.
Milwaukee School of Engineering (Milwaukee WI
Insulin Receptor
Team members: Selena Dickinson, Tori St. Martin, Kyra Gudgel and Arsalon Aminihajibashi
Faculty advisor: Jung, Lee, Ph.D
Research Mentor: George A. Wilkinson, Ph.D., Concordia University Wisconsin, Mequon WI
PDB ID: 3loh
Abstract
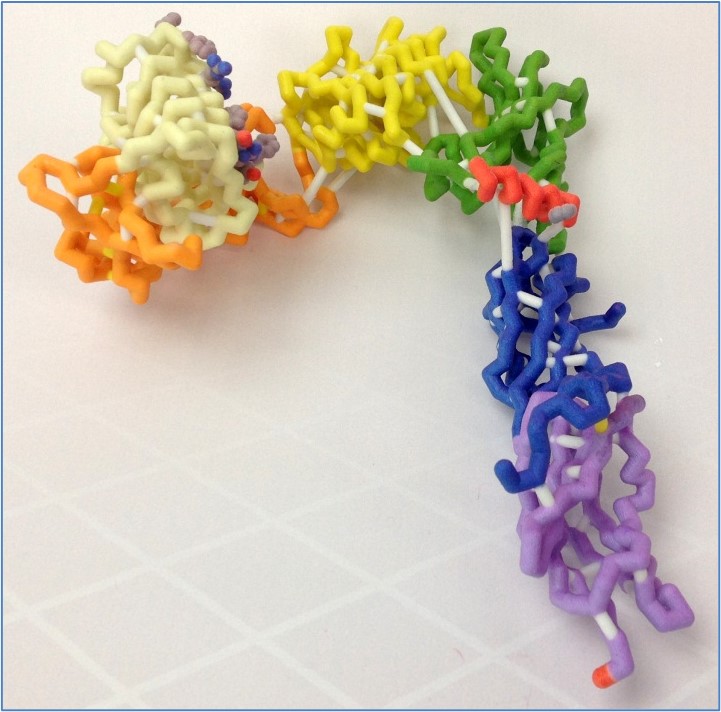 The human insulin receptor is a transmembrane protein found in skeletal muscle, fat tissue, and liver tissue, and its major function is to regulate glucose levels in the blood. The receptor exists as an upside-down V-shaped dimer, with each monomer cleavable to α and β chains. While the extracellular α-chain, made up of six structural domains, contains the insulin-binding domain in close proximity to its terminal helix (αCT), the intracellular β-chain is the kinase domain. Upon binding to insulin, the receptor undergoes dramatic conformational changes in the intracellular β-chain, initiating a cascade of signals for glucose uptake from the blood. Defects in the insulin-signaling cascade are directly associated with insulin resistance, a key feature of type 2 diabetes. We modeled the extracellular portion of the insulin receptor to better understand its three-dimensional structure in the presence or absence of bound insulin. Based on the PDB file 3LOH, we designed a three-dimensional physical model for the insulin-unbound of the extracellular portion of the monomeric insulin receptor, while MSOE’s other CREST team designed its insulin-bound structure using PDB file 3W14. The differences in the orientation of the insulin molecule between the two models illustrate potential conformational changes that may occur upon insulin binding. Also, although the 3LOH model does not show the protein as a dimer, the 3LOH protein has a more complete crystal structure for the chain it displays. It is interesting to note that the insulin receptor contains an unusually large (~40) number of disulfide bonds.
The human insulin receptor is a transmembrane protein found in skeletal muscle, fat tissue, and liver tissue, and its major function is to regulate glucose levels in the blood. The receptor exists as an upside-down V-shaped dimer, with each monomer cleavable to α and β chains. While the extracellular α-chain, made up of six structural domains, contains the insulin-binding domain in close proximity to its terminal helix (αCT), the intracellular β-chain is the kinase domain. Upon binding to insulin, the receptor undergoes dramatic conformational changes in the intracellular β-chain, initiating a cascade of signals for glucose uptake from the blood. Defects in the insulin-signaling cascade are directly associated with insulin resistance, a key feature of type 2 diabetes. We modeled the extracellular portion of the insulin receptor to better understand its three-dimensional structure in the presence or absence of bound insulin. Based on the PDB file 3LOH, we designed a three-dimensional physical model for the insulin-unbound of the extracellular portion of the monomeric insulin receptor, while MSOE’s other CREST team designed its insulin-bound structure using PDB file 3W14. The differences in the orientation of the insulin molecule between the two models illustrate potential conformational changes that may occur upon insulin binding. Also, although the 3LOH model does not show the protein as a dimer, the 3LOH protein has a more complete crystal structure for the chain it displays. It is interesting to note that the insulin receptor contains an unusually large (~40) number of disulfide bonds.
Mount Mary University (Milwaukee WI)
Argonaute-2 with associated miRNA
Team members: Monica Hernandez, DeAnn Kennedy and Molly Rose Rasper
Faculty advisors: Colleen Conway, Ph.D. and Maureen Leonard, Ph.D.
Research Mentor: Allison Abbott, Ph.D.; Marquette University
PDB ID: 4f3t
Abstract
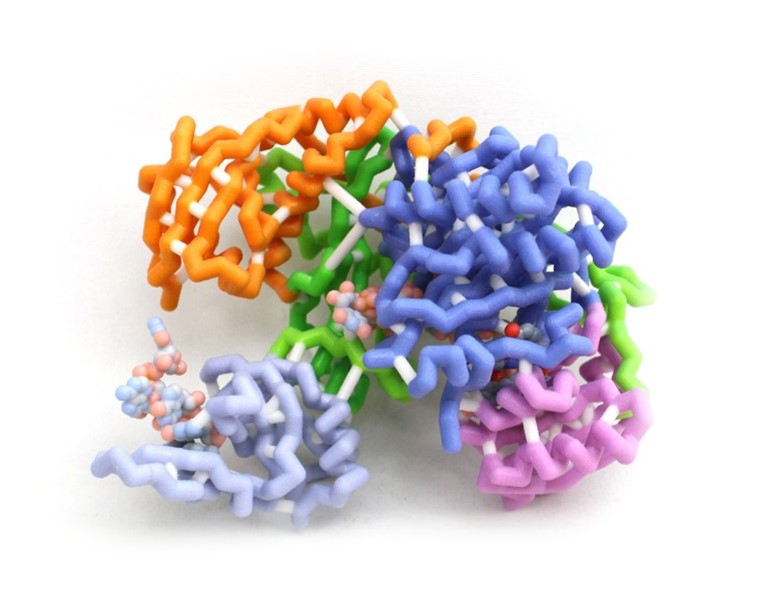 Development is a progressive program requiring precise amounts of proteins at specific times in the process to ensure proper form and function of the individual. This is accomplished by controlling gene expression among other mechanisms. Argonaute-2 (AGO-2) is a key part of the RNA induced silencing complex (RISC) used to prevent translation of mRNAs and is highly conserved in all domains of life. The RISC contains micro-RNAs (miRNAs) to bind specific mRNAs using a complementary seed sequence. AGO-2 then cleaves the mRNA as an RNase. This model highlights the amino acids that hold the miRNA in place within the RISC and the amino acids of the active site of AGO-2. The miRNA is held in place by positively charged amino acids interacting with the phosphates of the miRNA backbone to stabilize the molecule allowing complementation. This allows many different miRNAs to bind to AGO-2 regardless of the base sequence. The active site of AGO-2 contains amino residues typical of enzymes of the RNase H family. These residues hydrolyze the sugar-phosphate backbone of the mRNA. Silencing can be used to fine-tune the timing and dosage of important protein products. Failure of mRNA silencing prevents successful development.
Development is a progressive program requiring precise amounts of proteins at specific times in the process to ensure proper form and function of the individual. This is accomplished by controlling gene expression among other mechanisms. Argonaute-2 (AGO-2) is a key part of the RNA induced silencing complex (RISC) used to prevent translation of mRNAs and is highly conserved in all domains of life. The RISC contains micro-RNAs (miRNAs) to bind specific mRNAs using a complementary seed sequence. AGO-2 then cleaves the mRNA as an RNase. This model highlights the amino acids that hold the miRNA in place within the RISC and the amino acids of the active site of AGO-2. The miRNA is held in place by positively charged amino acids interacting with the phosphates of the miRNA backbone to stabilize the molecule allowing complementation. This allows many different miRNAs to bind to AGO-2 regardless of the base sequence. The active site of AGO-2 contains amino residues typical of enzymes of the RNase H family. These residues hydrolyze the sugar-phosphate backbone of the mRNA. Silencing can be used to fine-tune the timing and dosage of important protein products. Failure of mRNA silencing prevents successful development.
University of Wisconsin (Platteville WI)
Cyanovirin-N (CV-N)
Team members: Jessica McCollough and Katie Peterson
Faculty advisor: Sharon Klavins, Ph.D.
PDB ID: 2ezn
Abstract
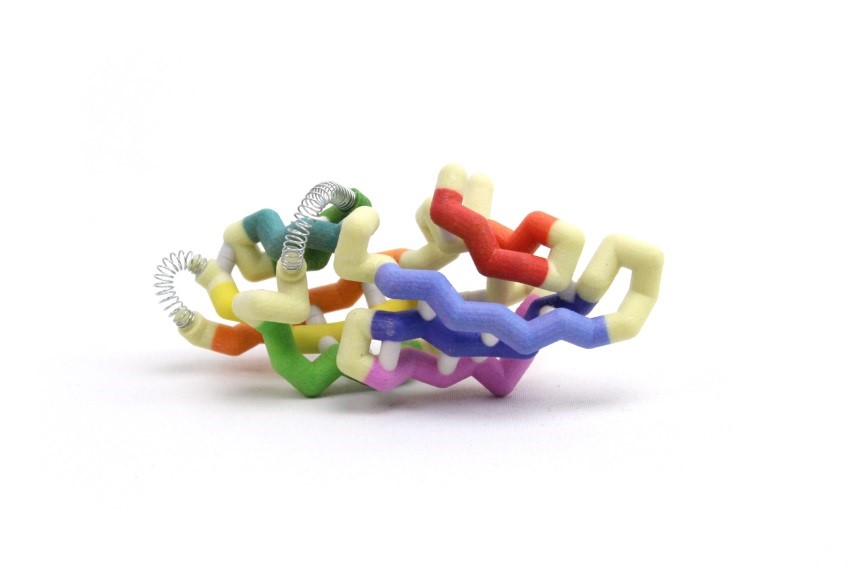 Cyanovirin-N (CV-N) is a protein in cyanobacteria that may provide a stable, durable, and inexpensive way to prevent HIV infections. CV-N attaches to a glycoprotein (gp120) on the surface of HIV, which prevents the virus from binding to uninfected cells. The CV-N monomer has a semi-ellipsoidal shape containing 10 beta (β) sheets and 3 alpha (α) helices. CV-N has a domain A and domain B which can form multimonomers through domain swapping, where one monomer exchanges an identical domain with another. The domains are divided by a primary hinge-loop region between the β5 and β6 strands, consisting of amino acids Gln50-Asn53 in the center of the 101 amino acid chain. A secondary hinge-loop region (Arg76-Gln78) between the β7 and β8 strands was recently identified and provides new information on how CV-N folds. The protein can fold, unfold, refold and take different folding paths to get to the desired quaternary structure. The hinges allow these folds, changing the torsion of the angles which directly influences the binding ability of the protein. In one folding pathway, the primary hinge flips the β6 strand up and backwards while the secondary hinge twists and straightens the β7 and β8 strands. The secondary hinge then swings upward, forming a three-stranded beta sheet. In the other pathway, the secondary hinge swings and creates the three-stranded sheet before the primary hinge swings the β6 strand upwards to form the semi-ellipsoidal monomer. These folding pathways are important in understanding the formation of quaternary structure.
Cyanovirin-N (CV-N) is a protein in cyanobacteria that may provide a stable, durable, and inexpensive way to prevent HIV infections. CV-N attaches to a glycoprotein (gp120) on the surface of HIV, which prevents the virus from binding to uninfected cells. The CV-N monomer has a semi-ellipsoidal shape containing 10 beta (β) sheets and 3 alpha (α) helices. CV-N has a domain A and domain B which can form multimonomers through domain swapping, where one monomer exchanges an identical domain with another. The domains are divided by a primary hinge-loop region between the β5 and β6 strands, consisting of amino acids Gln50-Asn53 in the center of the 101 amino acid chain. A secondary hinge-loop region (Arg76-Gln78) between the β7 and β8 strands was recently identified and provides new information on how CV-N folds. The protein can fold, unfold, refold and take different folding paths to get to the desired quaternary structure. The hinges allow these folds, changing the torsion of the angles which directly influences the binding ability of the protein. In one folding pathway, the primary hinge flips the β6 strand up and backwards while the secondary hinge twists and straightens the β7 and β8 strands. The secondary hinge then swings upward, forming a three-stranded beta sheet. In the other pathway, the secondary hinge swings and creates the three-stranded sheet before the primary hinge swings the β6 strand upwards to form the semi-ellipsoidal monomer. These folding pathways are important in understanding the formation of quaternary structure.
University of Wisconsin (Platteville WI)
Cyanovirin-N Homolog CrCVNH
Team members: Dugan Repass and Jessica Hauser
Faculty advisor: Sharon Klavins, Ph.D.
PDB ID: 2jzj
Abstract
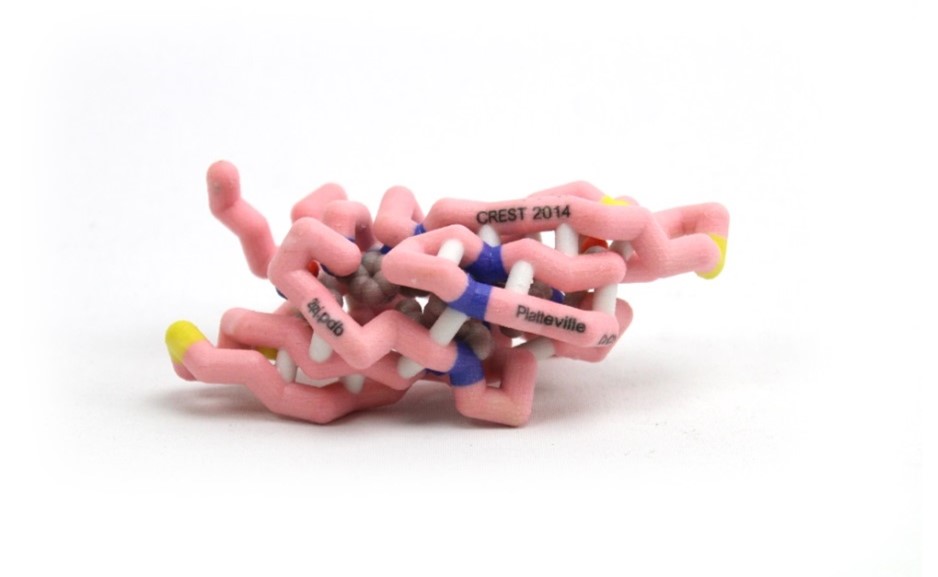 Cyanovirin-N (CV-N) is a protein being studied by scientists as a topical microbicide to prevent sexual transmission of human immunodeficiency virus (HIV). CV-N was isolated from the cyanobacterium, Nostoc ellipsosporum, and is composed of 101 amino acids making up two domains, and also has two disulfide bonds, critical for the fold and antiviral activity. Ceratopteris richardii is a fern from which CrCVNH, a homolog to CV-N, was isolated and shows a similar overall structure that resembles CV-N, although with noteworthy differences. There are 10 conserved residues between the two that play a key role in structural support and in carbohydrate binding. In addition, there are four glycines (28, 46, 79, and 97) and two asparagines (43 and 94) that are conserved in both CV-N and CrCVNH which may play vital roles in determining carbohydrate specificity of the members of the CVNH family. Despite predicted structural similarities between the homologs, one difference is that CrCVNH has an additional 12 residues at the end of the amino acid chain. CrCVNH has an additional disulfide bond between Cys3 and Cys102, which may help maintain protein form and function. Other differences are located in the sugar binding pockets of the proteins, which may explain the significantly lower anti-viral activity of CrCVNH when compared to CV-N.
Cyanovirin-N (CV-N) is a protein being studied by scientists as a topical microbicide to prevent sexual transmission of human immunodeficiency virus (HIV). CV-N was isolated from the cyanobacterium, Nostoc ellipsosporum, and is composed of 101 amino acids making up two domains, and also has two disulfide bonds, critical for the fold and antiviral activity. Ceratopteris richardii is a fern from which CrCVNH, a homolog to CV-N, was isolated and shows a similar overall structure that resembles CV-N, although with noteworthy differences. There are 10 conserved residues between the two that play a key role in structural support and in carbohydrate binding. In addition, there are four glycines (28, 46, 79, and 97) and two asparagines (43 and 94) that are conserved in both CV-N and CrCVNH which may play vital roles in determining carbohydrate specificity of the members of the CVNH family. Despite predicted structural similarities between the homologs, one difference is that CrCVNH has an additional 12 residues at the end of the amino acid chain. CrCVNH has an additional disulfide bond between Cys3 and Cys102, which may help maintain protein form and function. Other differences are located in the sugar binding pockets of the proteins, which may explain the significantly lower anti-viral activity of CrCVNH when compared to CV-N.
University of Wisconsin (Platteville WI)
Five Site Mutant P51G-m4-CVN: Understanding Antiviral HIV Properties Involving the Protein gp120
Team members: Audra Krusz, Kayla Koltemann and Taras Koshyk
Faculty advisor: Sharon Klavins, Ph.D.
PDB ID: 2z21
Abstract
 Cyanovirin-N (CV-N) is a protein found in cyanobacteria known to have antiviral properties against human immunodeficiency virus (HIV). CV-N binds to carbohydrates on the viral surface envelope protein gp120 and prevents fusion and transmission of HIV to the host cell, possibly leading to a preventative for the sexually transmitted virus. There are two domains, A and B, each consisting of approximately 50 amino acids and containing a single carbohydrate binding site. Scientists altered five sites on the wild type CV-N producing the mutant P51G-m4-CVN. The mutations, specifically m4 consisting of 4 amino acid changes, restricted domain A so only the binding site on domain B was active. Domain B had the same capability of binding to gp120 as the wild type CV-N. However, the mutated form of CV-N had an overall decrease in binding affinity to the gp120 protein and showed no antiviral activity. In the wild type CV-N there is a rare cis-peptide bond between gly51 and ser52. The P51G mutation changes gly51 to pro51, which is significant because in the wild type there is a hinge region from gln50 to asn53 which allows the protein to make conformational changes. The P51G mutation stabilizes CVN to crystallize exclusively as a monomer by changing the peptide bond in the hinge region. These mutations are significant as they show that whether in monomer or dimer form there must be two active sites on CV-N to bind to gp120.
Cyanovirin-N (CV-N) is a protein found in cyanobacteria known to have antiviral properties against human immunodeficiency virus (HIV). CV-N binds to carbohydrates on the viral surface envelope protein gp120 and prevents fusion and transmission of HIV to the host cell, possibly leading to a preventative for the sexually transmitted virus. There are two domains, A and B, each consisting of approximately 50 amino acids and containing a single carbohydrate binding site. Scientists altered five sites on the wild type CV-N producing the mutant P51G-m4-CVN. The mutations, specifically m4 consisting of 4 amino acid changes, restricted domain A so only the binding site on domain B was active. Domain B had the same capability of binding to gp120 as the wild type CV-N. However, the mutated form of CV-N had an overall decrease in binding affinity to the gp120 protein and showed no antiviral activity. In the wild type CV-N there is a rare cis-peptide bond between gly51 and ser52. The P51G mutation changes gly51 to pro51, which is significant because in the wild type there is a hinge region from gln50 to asn53 which allows the protein to make conformational changes. The P51G mutation stabilizes CVN to crystallize exclusively as a monomer by changing the peptide bond in the hinge region. These mutations are significant as they show that whether in monomer or dimer form there must be two active sites on CV-N to bind to gp120.
University of Wisconsin (Platteville WI)
Cyanovirin-N (CV-N)
Team members: Mollie Williams, Jennifer McCormick, Jillian Cassel
Faculty advisor: Sharon Klavins, Ph.D.
PDB ID: 3gxz
Abstract
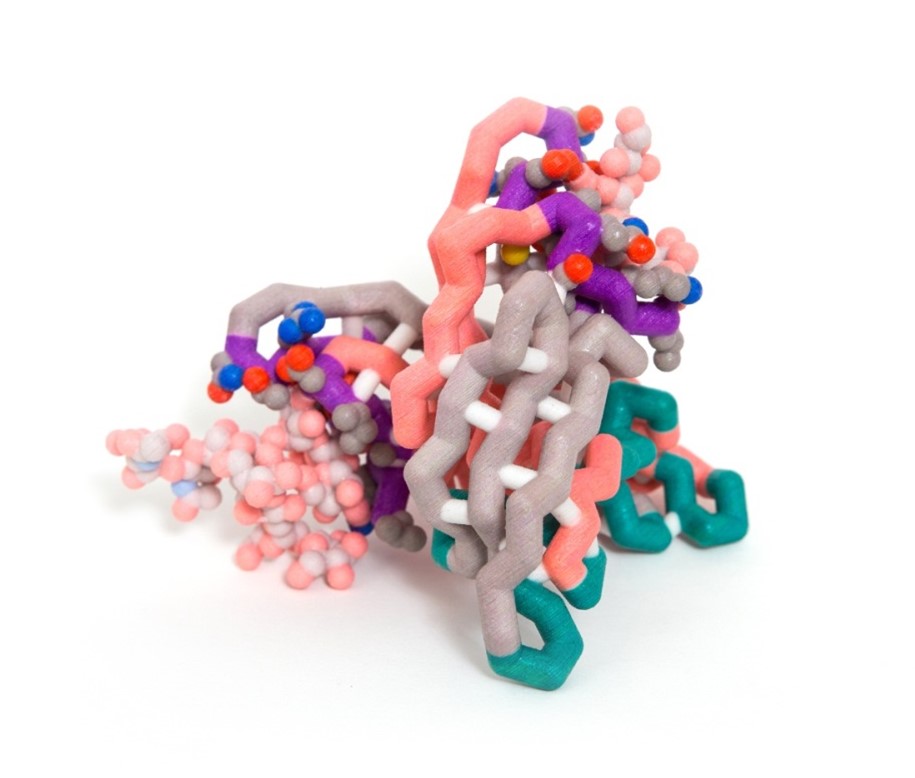 In order for a virus to reproduce, it needs to insert its own genetic material into a host; however certain proteins can prevent viruses from invading host cells. Cyanovirin-N (CVN) is a protein originally isolated from cyanobacteria that shows anti-HIV properties along with antiviral activity against Ebola, SIV, Influenza, and Hepatitis-C. Gp120 is a glycoprotein located on the outer envelope of the HIV virion. The surface of gp120 is covered with carbohydrates, which is critical to the binding of CVN to the glycoprotein. The CVN dimer is composed of β-sheets and loops that form binding sites with different affinities for carbohydrates on the virus envelope surface. The primary (high affinity) binding site lies near a hinge region and consists of residues 41-44, 50-56, and 74-78. The secondary (low affinity) binding site lies on the opposite sides of the dimer and consists of residues 1-7, 22-26, and 92-95. The secondary site is not influenced by the changes of the hinge region. The model of this molecule highlights both binding sites but shows the carbohydrates binding only at the secondary site. More than one binding site must be functional in order for CVN to have antiviral activity. CVN is not a cure for HIV and other viruses; however, it may have the potential to be developed into a microbicide to prevent HIV transmission and infection.
In order for a virus to reproduce, it needs to insert its own genetic material into a host; however certain proteins can prevent viruses from invading host cells. Cyanovirin-N (CVN) is a protein originally isolated from cyanobacteria that shows anti-HIV properties along with antiviral activity against Ebola, SIV, Influenza, and Hepatitis-C. Gp120 is a glycoprotein located on the outer envelope of the HIV virion. The surface of gp120 is covered with carbohydrates, which is critical to the binding of CVN to the glycoprotein. The CVN dimer is composed of β-sheets and loops that form binding sites with different affinities for carbohydrates on the virus envelope surface. The primary (high affinity) binding site lies near a hinge region and consists of residues 41-44, 50-56, and 74-78. The secondary (low affinity) binding site lies on the opposite sides of the dimer and consists of residues 1-7, 22-26, and 92-95. The secondary site is not influenced by the changes of the hinge region. The model of this molecule highlights both binding sites but shows the carbohydrates binding only at the secondary site. More than one binding site must be functional in order for CVN to have antiviral activity. CVN is not a cure for HIV and other viruses; however, it may have the potential to be developed into a microbicide to prevent HIV transmission and infection.
Saint Leo University (Saint Leo FL)
Protein Kinase C δ ll (PKCδ II)
Team members: Amanda Morris, Ann Anselme and Antonio Roki
Faculty advisor: Audrey Shor, Ph.D.
Research Mentors: Niketa Patel, Ph.D.and Jeremy Prokop, Medical College of Wisconsin
Theoretical Model:RaptorX
Abstract
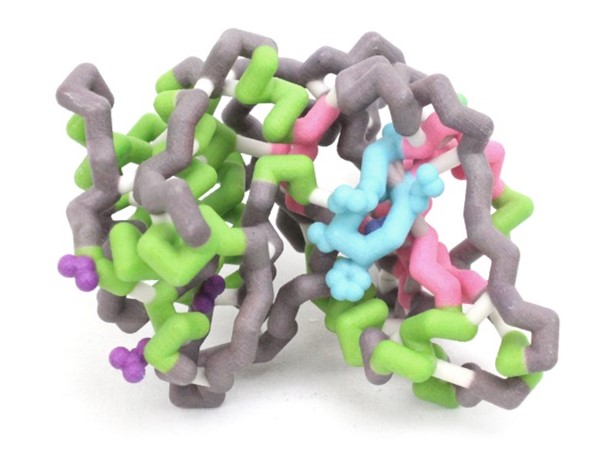 Protein kinase C δ (PKCδ) has been shown to have dual functions as a regulator of both apoptosis and cellular survival due to alternative splicing. One splice variant, PKCδ II, is integral in promoting cell survival. The pro-apoptotic protein, BCL-2 antagonist of cell death (BAD), complexes with and inhibits the function of pro-survival proteins. Upon phosphorylation at Ser-136, BAD disassociates from these complexes, conferring survival. Overexpression of PKCδII coincides with increased BAD phosphorylation. Although the exact mechanism is unknown, we believe PKCδII enhances this phosphorylation via direct interactions with BAD. We aim to elucidate the amino acid residues of PKCδII that interact with BAD. Based on sequence analyses and computational simulations, we propose the interactions that complex PKCδII with BAD. The amphipathic groove (Lys-49, Val-176, Leu-220) of 14-3-3 has previously been demonstrated to interact with BAD. Upon alignment of the 14-3-3 and PKCδII amino acid sequences, it was noted that the residues that comprise the amphipathic groove may be conserved and also facilitate binding of PKCδII with BAD. The PKCδII residues Gln-545, Val-547, and Val-586 may enable the formation of this complex. A hypothetical model of PKCδII and BAD has aided in visualizing this interaction. The proposed protein structures were generated using two servers; RaptorX for protein threading structure predictions and QUARK for ab initio methods. Molecular dynamic simulations were completed to test hydrophobic collapse of the theoretical structures using YASARA. Subsequent site-directed mutagenesis will further solidify our understanding of this protein complex. These proposed interactions provide evidence to suggest that BAD complexes directly with PKCδII in a conserved amphipathic groove, thereby leading to increased survivability.
Protein kinase C δ (PKCδ) has been shown to have dual functions as a regulator of both apoptosis and cellular survival due to alternative splicing. One splice variant, PKCδ II, is integral in promoting cell survival. The pro-apoptotic protein, BCL-2 antagonist of cell death (BAD), complexes with and inhibits the function of pro-survival proteins. Upon phosphorylation at Ser-136, BAD disassociates from these complexes, conferring survival. Overexpression of PKCδII coincides with increased BAD phosphorylation. Although the exact mechanism is unknown, we believe PKCδII enhances this phosphorylation via direct interactions with BAD. We aim to elucidate the amino acid residues of PKCδII that interact with BAD. Based on sequence analyses and computational simulations, we propose the interactions that complex PKCδII with BAD. The amphipathic groove (Lys-49, Val-176, Leu-220) of 14-3-3 has previously been demonstrated to interact with BAD. Upon alignment of the 14-3-3 and PKCδII amino acid sequences, it was noted that the residues that comprise the amphipathic groove may be conserved and also facilitate binding of PKCδII with BAD. The PKCδII residues Gln-545, Val-547, and Val-586 may enable the formation of this complex. A hypothetical model of PKCδII and BAD has aided in visualizing this interaction. The proposed protein structures were generated using two servers; RaptorX for protein threading structure predictions and QUARK for ab initio methods. Molecular dynamic simulations were completed to test hydrophobic collapse of the theoretical structures using YASARA. Subsequent site-directed mutagenesis will further solidify our understanding of this protein complex. These proposed interactions provide evidence to suggest that BAD complexes directly with PKCδII in a conserved amphipathic groove, thereby leading to increased survivability.
Saint Leo University (Saint Leo FL)
BCL-2 Antagonist of Cell Death (BAD)
Team members: Amanda Morris, Ann Anselme and Antonio Roki
Faculty advisor: Audrey Shor, Ph.D.
Research Mentor: Niketa Patel, University of South Florida
Theoretical Model: QUARK
Abstract
 Protein kinase C δ (PKCδ) has been shown to have dual functions as a regulator of both apoptosis and cellular survival due to alternative splicing. One splice variant, PKCδ II, is integral in promoting cell survival. The pro-apoptotic protein, BCL-2 antagonist of cell death (BAD), complexes with and inhibits the function of pro-survival proteins. Upon phosphorylation at Ser-136, BAD disassociates from these complexes, conferring survival. Overexpression of PKCδII coincides with increased BAD phosphorylation. Although the exact mechanism is unknown, we believe PKCδII enhances this phosphorylation via direct interactions with BAD. We aim to elucidate the amino acid residues of PKCδII that interact with BAD. Based on sequence analyses and computational simulations,.we propose the interactions that complex PKCδII with BAD. The amphipathic groove (Lys-49, Val-176, Leu-220) of 14-3-3 has previously been demonstrated to interact with BAD. Upon alignment of the 14-3-3 and PKCδII amino acid sequences, it was noted that the residues that comprise the amphipathic groove may be conserved and also facilitate binding of PKCδII with BAD. The PKCδII residues Gln-545, Val-547, and Val-586 may enable the formation of this complex. A hypothetical model of PKCδII and BAD has aided in visualizing this interaction. The proposed protein structures were generated using two servers; RaptorX for protein threading structure predictions and QUARK for ab initio methods. Molecular dynamic simulations were completed to test hydrophobic collapse of the theoretical structures using YASARA. Subsequent site-directed mutagenesis will further solidify our understanding of this protein complex. These proposed interactions provide evidence to suggest that BAD complexes directly with PKCδII in a conserved amphipathic groove, thereby leading to increased survivability.
Protein kinase C δ (PKCδ) has been shown to have dual functions as a regulator of both apoptosis and cellular survival due to alternative splicing. One splice variant, PKCδ II, is integral in promoting cell survival. The pro-apoptotic protein, BCL-2 antagonist of cell death (BAD), complexes with and inhibits the function of pro-survival proteins. Upon phosphorylation at Ser-136, BAD disassociates from these complexes, conferring survival. Overexpression of PKCδII coincides with increased BAD phosphorylation. Although the exact mechanism is unknown, we believe PKCδII enhances this phosphorylation via direct interactions with BAD. We aim to elucidate the amino acid residues of PKCδII that interact with BAD. Based on sequence analyses and computational simulations,.we propose the interactions that complex PKCδII with BAD. The amphipathic groove (Lys-49, Val-176, Leu-220) of 14-3-3 has previously been demonstrated to interact with BAD. Upon alignment of the 14-3-3 and PKCδII amino acid sequences, it was noted that the residues that comprise the amphipathic groove may be conserved and also facilitate binding of PKCδII with BAD. The PKCδII residues Gln-545, Val-547, and Val-586 may enable the formation of this complex. A hypothetical model of PKCδII and BAD has aided in visualizing this interaction. The proposed protein structures were generated using two servers; RaptorX for protein threading structure predictions and QUARK for ab initio methods. Molecular dynamic simulations were completed to test hydrophobic collapse of the theoretical structures using YASARA. Subsequent site-directed mutagenesis will further solidify our understanding of this protein complex. These proposed interactions provide evidence to suggest that BAD complexes directly with PKCδII in a conserved amphipathic groove, thereby leading to increased survivability.