
Getting Started in Jmol
This Jmol Exploration was created using the Jmol Exploration Webpage Creator from the MSOE Center for BioMolecular Modeling.
Physical models of proteins are powerful tools that can be used synergistically with computer visualization tools to explore protein structure and function. Although it is interesting to explore models and visualizations created by others, it is much more engaging to create your own!
A number of molecular visualization programs are on the market; we have selected to use Jmol, which is a free open source molecular visualization program used by students, educators and researchers. These online resources will provide the tools needed to create physical models using 3-D printing technologies, as well as Jmol animations for online Jmol Explorations or electronic posters.
Welcome to the amazing world of proteins – and the fascinating stories that each one can tell!
Jmol runs on a Java platform and therefore functions equally well in a PC or Mac environment. Because Jmol is freeware and an open source program, it can be obtained from many locations. For total functionality in designing a structure to be built on a 3-D printer in the CBM, check the website every September to get the current version of Jmol that we will be using for the school year. Note that files created in older versions can be viewed in more recent versions, but files created in more recent versions may not open in older versions of Jmol.
We suggest that you download Jmol directly from the CBM website to ensure you are using the correct version for creating physical models. You may click the link below to download Jmol.
You need to have Java on your computer to run the desktop version of Jmol. Most computers will already have Java installed, but if you do not, you can download it for free from https://java.com/en/download/.
Jmol Version 14.31.36 (Mac or PC) (Zipped folder with Jmol.jar and Jmol.bat; use Jmol.jar)
Jmol does not need to be 'installed', so once the Jmol file is in place on your computer, the program will be ready to use. Click on the Jmol.jar file to start Jmol on your computer.
You may save the Jmol file you download in any convenient location on your computer. Jmol does not need to be 'installed', so once these files are in place on your computer, the program will be ready to use. Click on the Jmol file to start Jmol on your computer. Files you save in Jmol will be saved in the same file location as the Jmol file.
Molecular structures can be determined through the use of X-ray Crystallography, Nuclear Magnetic Resonance (NMR), or computer algorithm-based calculations. Once a structure has been determined, each atom in the structure is assigned an (X,Y,Z) coordinate to mark its location in 3-dimensional space. These coordinates are then stored in a file called a Protein Data Bank (PDB) file. Molecular visualization software, such as Jmol, can then use the coordinates stored in a PDB file to create an interactive 3-dimensional visualization of a molecular structure.
The most common source of free PDB files of macromolecules is the RCSB Protein Data Bank website at
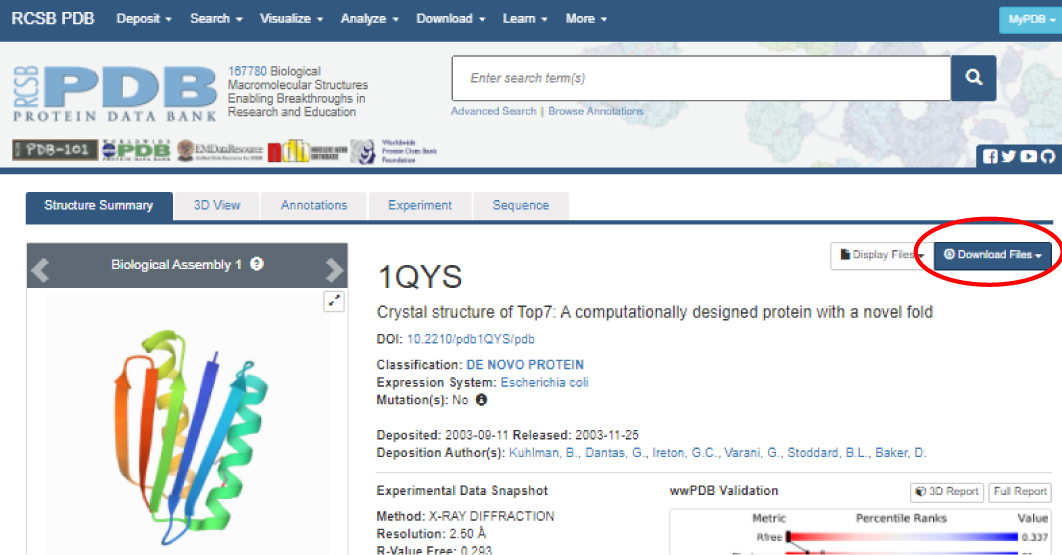
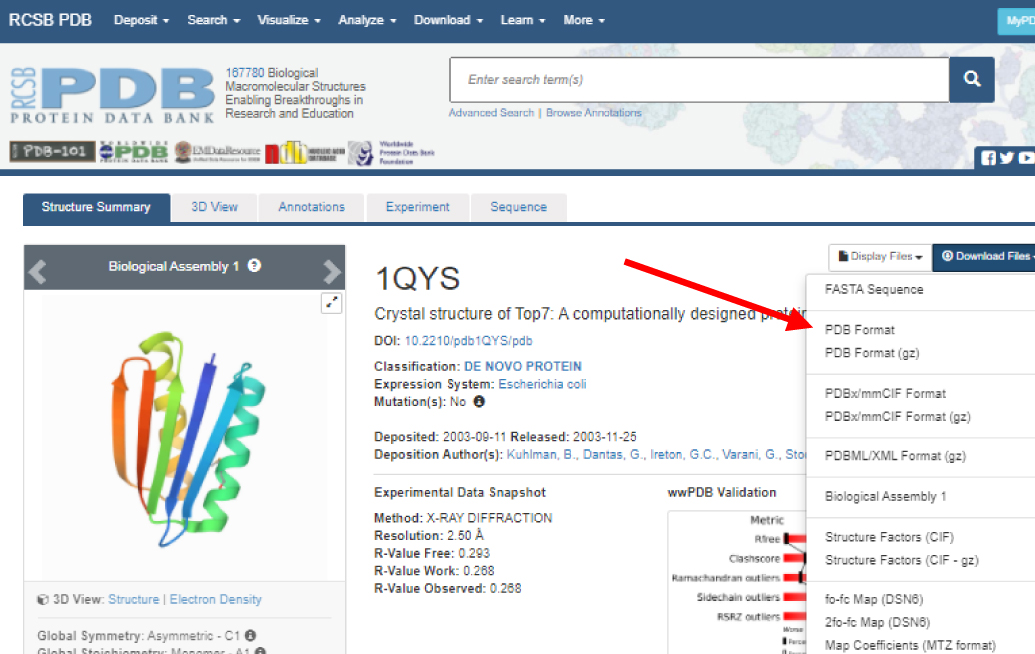
Note: It is a good idea to create a new folder for each molecule you explore to organize all PDB files, images, articles and other related work. (See the 'Saving Your Work and Organizing Your Files Training Guide' for more information.)
If you don't need additional information about the PDB file or don't wish to save a copy of the file on your computer, you can
'call up' the file in Jmol without downloading. See Saving and Opening Files in Jmol for more details.
To launch Jmol, click on the 'Jmol.jar'file. Note: it should have an icon similar to one of the images below.


Quick tip: You might wish to create a shortcut to the 'Jmol.jar' file on your desktop and in each of your working folders so you can easily find and launch Jmol.
Once launched, the Jmol Display Window will appear. The Jmol Display Window will show the 3-dimensional structure of the PDB structures you load and design. You can adjust the size of this window by grabbing an edge or corner of the screen with your mouse then clicking and dragging to adjust size. Most people prefer to have this window to occupy half to two-thirds of your screen.
The Jmol Console is used to input commands. To access the Console, right click anywhere in the Display Window to bring up the Display Window Menu. In this menu, click on the button 'Console' which will open the Jmol Console.
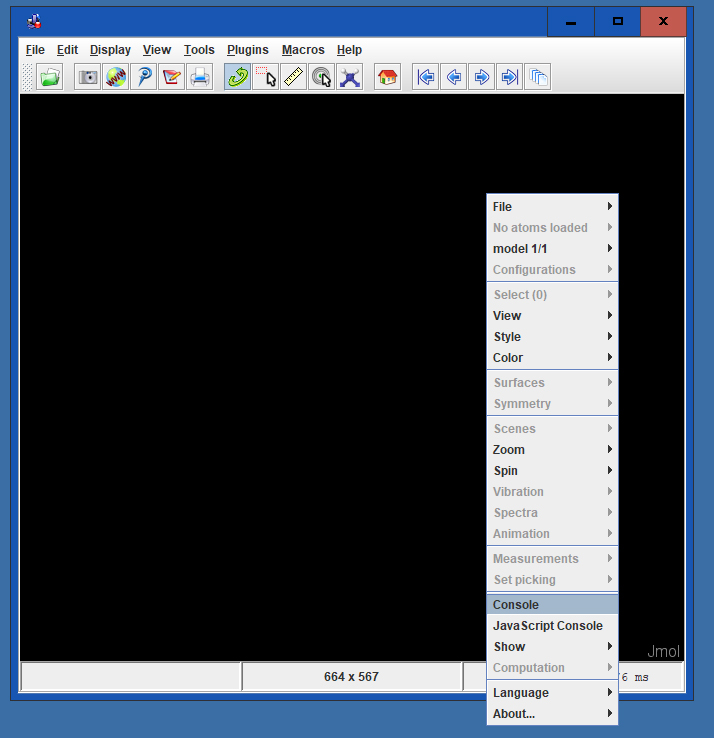
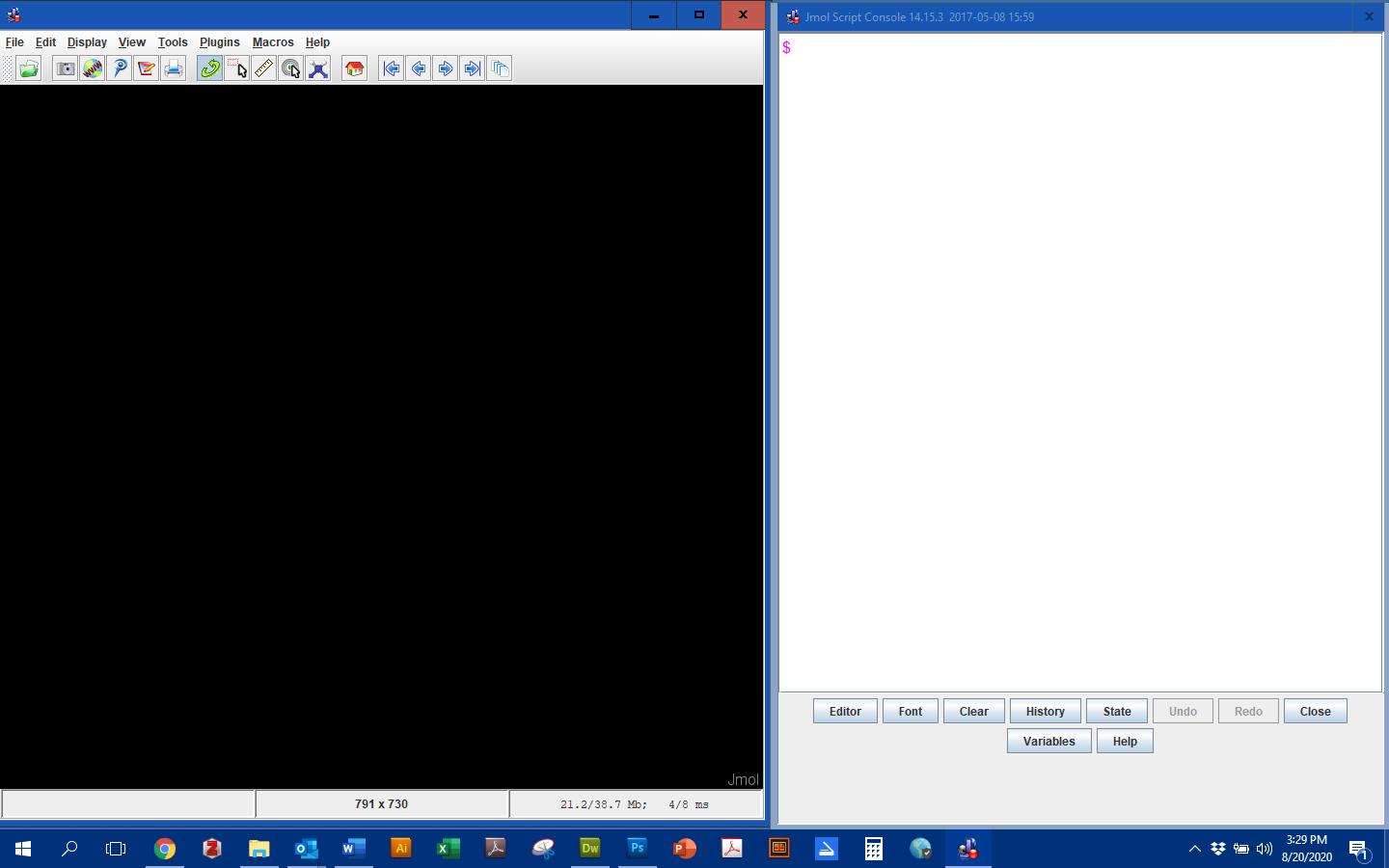
Because the Console and Display Windows will almost always be used in unison, arrange the windows so that they do not overlap and both fit comfortably on your screen. Most people prefer a larger Display Window.To move the windows, click and drag in the header space.
There are two options for opening a PDB file.
Option 1: Open the folder that contains your PDB file. Click and drag your PDB file into the Display Window. Note: you may have to arrange the Jmol window and folder window in such a way that you can see both at the same time in order to click and drag the PDB file.
Option 2: If you have Internet access and don't want to store PDB files on your computer, in the console, enter the command:
load=xxx, where xxxx represents the four character PDB code.
To load Top7 using 1qys.pdb, use the command:
load=1qys
Depending on the PDB file, the structure may appear in one of several formats:
You'll learn how to change the appearance of the structure in other tutorials.
Once you have opened your PDB file, the structure should appear in your Jmol Display Window. Click the button below to see what your Jmol Window might look like. (This is a ball and stick rendering.)
Top 7 PDB ID: 1qysYou can now rotate, relocate or zoom in on the molecule in the Display Window.
To rotate the molecule within the X-Y axes on the computer screen,
To move ('translate') the molecule,
To zoom in or out,
To barrel roll (rotate the molecule on the Z axis),